动物源性样品制备国内外有没有相关的规范,指南??尤其是分样,破碎。比如,来一份样品,是取可食部分进行制样?还是整个都作为检测对象?
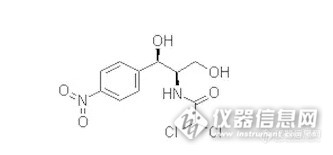
随着现代化市场经济的迅猛发展,人们的生活条件得到了改善,对动物源性食品的需求也越来越多,其安全问题也随之越来越重要,其中,氯霉素残留是较为突出的因素之一。那么,氯霉素是什么?其在动物源性食品中的现状又是怎样的呢?动物源性食品 动物源性食品是指来源于动物,可供人类食用的动物产品,包括肉、脂肪、脏器、血液、蛋、奶等。随着国民经济的发展,我国城乡居民的生活水平和健康消费观念的提高,动物源性食品在我国居民食品结构中占有比例越来越大,需求量不断增大,拉动了畜牧业的快速发展。然而,近几年发生的“瘦肉精”“掺假羊肉”“病死猪肉”“三鹿婴幼儿奶粉”等事件使消费者谈“肉”色变,动物源性食品的安全状况已成为当今广受关注的社会话题。氯霉素http://ng1.17img.cn/bbsfiles/images/2015/08/201508251507_562769_2984502_3.jpg 氯霉素为白色针状或微带黄绿色的针状、长片状结晶或结晶性粉末,味苦,易溶于甲醇、乙醇、丙酮,微溶于水,干燥时稳定。氯霉素的化学结构含有对硝基苯基、丙二醇与二氯乙酰胺三个部分, 因其分子中含有一个不游离的氯,故命名氯霉素,其抗菌活性主要与丙二醇有关。 氯霉素是由委内瑞拉链丝菌产生的一种抑菌性广谱抗生素,它通过与核糖体的50s亚单位结合而抑制细菌蛋白质的合成。对革兰氏阳性菌和革兰氏阴性菌均有较好的抑制作用,对立克次体、衣原体也有抑制作用。因其高效廉价,曾在畜牧业中广为应用。然而由于其对造血系统有严重的不良反应,且细菌对氯霉素有发展缓慢的耐药性,所以对其临床应用已经做出严格控制。动物源性食品中氯霉素的现状 国际上对动物源性农产品的兽药残留问题亦广泛关注,世界上许多国家禁止氯霉素使用于生产食品动物,并规定了其在畜产品中的最高残留限量。欧盟、美国等均在其相关法规中规定氯霉素的残留为“零容许量” ,即不得检出。 我国农业部已将氯霉素从2000年版的《中国兽药典》中删除,作为禁用药品。在2002年底的农业部第235号公告《动物源性食品中兽药最高残留限量》中也明确规定氯霉素禁止使用,在动物性食品中不得检出。现在,氯霉素是动物性农产品的必检指标。动物源性食品中氯霉素的检测 关于动物源性食品中氯霉素的检测,主要使用的标准方法有:GB/T22338-2008《动物源性食品中氯霉素类药物残留量测定》、GB/T20756-2006《可食动物肌肉、肝脏和水产品中氯霉素、甲砜霉素和氟苯尼考残留量的测定 液相色谱-串联质谱法》、GB29688-2013《食品安全国家标准 牛奶中氯霉素残留量的测定 液相色谱-串联质谱法》、GB/T18932.19-2003《蜂蜜中氯霉素残留量的测定方法 液相色谱-串联质谱法》等。食品生产企业和销售单位应把动物源性食品中氯霉素的检测作为原料质控和进货采购验证的一项重要指标进行管理,要求供应商出具相关检测报告,必要时可委托有资质的第三方检测机构进行检测。
1) SPE 方法 订货号:C2160107-304 固定相: Thermo HyperSep Retain-CX 柱体积: 3mL 固定相重量:200mg 酶解: 动物源性样品2g (精确到0.01g)于50mL离心管中,加入0.2 mol/L乙酸铵溶液(pH 5.2)10mL,然后加入β-盐酸葡萄糖醛苷酶/芳基硫酸酯酶40μL,涡旋混匀3 min,于37℃下水浴避光振荡16h。 提取:样品酶解后放置至室温,涡旋混匀3 min,高速离心10min,取出上清液,加入1mol/L高氯酸溶液1mL,涡旋,混匀,高速离心10min后,转移上清液至另一50mL离心管内。 活化:3mL甲醇,3mL水,3mL 0.5mol/L高氯酸 上样:样品 清洗:3 mL水,3mL甲醇,柱子抽干 洗脱:3mL5%氨水甲醇溶液 2)LC/MS 方法 定货号:25005-152130 色谱柱:Hypersil Gold,5μm,2.1×150mm 流动相:A:水(5mM乙酸铵)B:甲醇,梯度洗脱: http://s01.yizimg.com/images/news/159/6defed78881b410beacb85d03e158550.jpg 进样量:10μL 流速:250μL/min MS条件:电喷雾电离源(ESI),正离子模式 选择反应监控(SRM)扫描模式 喷雾电压:4500V 离子传输管温度:350℃
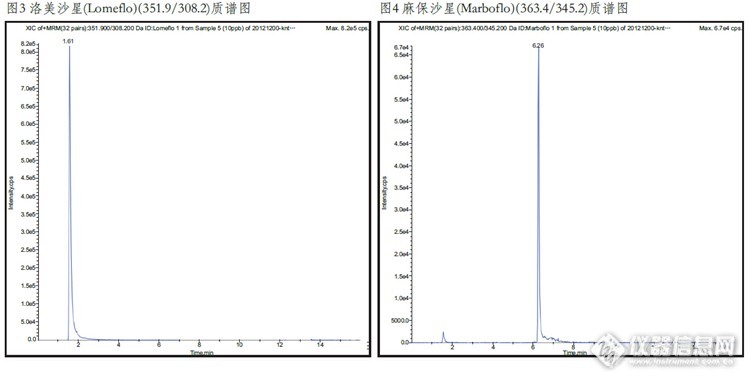
动物源性食品中喹诺酮类物质检测的固相萃取方法 一、实验目的本研究利用固相萃取法作为猪肉样品的前处理方法,LC-MS/MS法作为检测手段。该方法可简化猪肉样品的前处 理过程,节省有机溶剂的使用,操作简便。二、实验目标物三种喹诺酮类药物:依诺沙星(CAS:74011-58-8;84294-96-2),洛美沙星(CAS:98079-51-7),麻保沙星(CAS:115550-35-1)。三、应用范围本方法适用于动物源食品中喹诺酮类药物LC-MS/MS检测及确证。四、参考标准进出口行业标准《SN/T1751.2-2007动物源性食品中喹诺酮类药物残留检测方法;第2部分:液相色谱-质谱/质 谱法》。五、实验材料Biocomma® PolybaseTM HLB固相萃取柱3mL/60mg。六、实验方法1、样品提取称取均质试样5.0g试样(精确至0.01g)于50mL离心管中,加入20mL乙腈,涡旋混合1min,超声提取10min,10000r/min离心,提取三次,合并上清液。2、SPE柱净化(1)活化:加入6mL甲醇,6mL水活化。(2)上样和洗脱:当溶剂液面到达柱吸附层表面时,立即倒入上述待净化溶液,以2mL/min-3mL/min的速度过 柱,弃去滤液,用2mL5%甲醇水溶液淋洗,弃去淋洗液,将小柱抽干,在用6mL甲醇洗脱并收集洗脱液。(3)浓缩定容:50℃缓慢氮气流条件下吹至近干,用1mL0.2%甲酸水溶液溶解,1000r/min旋涡混合1min[color
动物源性食品中链霉素检测,使用国标GB/T 21323-2007,检测回收率不是很高,有没有其他改进的建议,谢谢
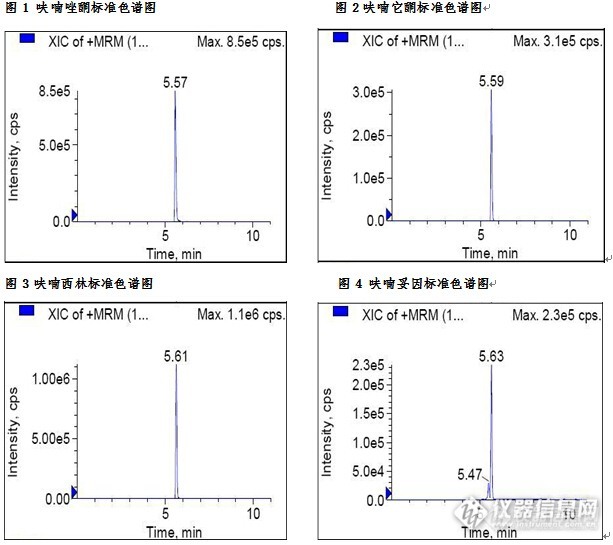
动物源性食品中硝基呋喃类药物检测的固相萃取方法一、实验目的本实验利用固相萃取法作为样品的前处理方法,LC-MS/MS法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂用量。二、实验目标物呋喃唑酮(CAS:67-45-8),呋喃它酮(CAS:139-91-3),呋喃西林(CAS:59-87-0),呋喃妥因(CAS:67-20-9)。三、应用范围本方法适用于动物源性食品中硝基呋喃类药物的LC-MS/MS检测及确证。四、参考文献 推荐性国家标准《GB/T 21311-2007 动物源性食品中硝基呋喃类药物代谢物残留量检测方法 高效液相色谱-串联质谱法》。五、实验材料 C8/SAX固相萃取柱200mg/6mL。六、实验方法1、样品前处理 将样品组织搅碎、均质。精确称取约1g(精确到0.01g)样品于15mL带螺盖的离心管中,加入1mL水和8mL甲醇,涡旋混合均匀,2000r/min离心3min(15℃),弃去上清液;加入8mL乙醇,涡旋混合均匀,2000r/min离心3min(15℃),弃去上清液;加入8mL乙酸乙酯,涡旋混合均匀,2000r/min离心3min(15℃),弃去上清液。2、水解和衍生化向质控和样品同待测样品中加入5mL 0.2mol/L的盐酸水溶液、100μL衍生化试剂、以及100μL内标工作溶液(20μg/L)和的混合标准溶液(10μg/L)。盖好盖子,充分振荡混匀,然后放入空气浴摇床,在37℃,200r/min条件下衍生化16h(过夜)。3、样品提取 加入0.3M的磷酸钠水溶液500μL。用试管滴加10mol/L的氢氧化钠溶液,涡旋混合均匀,用精密pH试纸调节pH值至7.0—7.2。9500r/min 离心10min,取上清液。4、SPE柱净化(1)活化:依次以5mL甲醇和5mL纯水预处理。(2)洗脱:上清液全部过柱,流速控制在约每秒1滴。依次以5mL水和5mL 50%甲醇/水溶液淋洗柱子。淋洗液完全通过小柱后,至少抽真空5min。以4mL 4%的甲醇氨洗脱,洗脱液用15mL试管收集。(3)浓缩定容:40℃氮气吹干。残渣以0.05%甲酸/甲醇溶液(9:1,v/v)溶解。溶解液以0.22μm的水相滤膜过滤,滤液可直接用于LC-MS/MS分析。5、LC-MS/MS条件 液相色谱-质谱/质谱仪 色谱柱:C18柱:150mm×2.1mm,2.0μm,或相当者 流动相:甲醇-5mM乙酸铵七、实验结果1、添加回收结果 向样品中加入不同水平的四环素类药物,回收率结果如下:(见表1)表1 动物组织中四环素类药物添加回收结果 样品名称 化合物名称 添加水平(ng/mL) 回收率(%) 猪肉 呋喃唑酮 50 80.75 100 82.58 呋喃它酮 50 89.74 100 90.88 呋喃西林 50 92.74 100 91.28 呋喃妥因 50 95.63 100 96.94 2、 空白样品添加农药残留物色谱图 http://ng1.17img.cn/bbsfiles/images/2015/08/201508141653_560805_3310_3.jpg
PCR技术在食品动物源性检测中的应用注:全文见资料中心。
鄙人最近接到一个比较棘手的检测项目,做动物源性食品中的粘杆菌素,但相关的标准或是文献很是少,我是用了博纳艾杰尔的固相萃取柱处理,然后LC/MS进行检测,结果回收率还可以接受,但谱图不是很好。浓度低时谱图容易出现分叉或是肩峰,而且做一些样品后,峰形变的更不好。所以恳请哪位高手可以赐教,如何解决此类问题,是前处理的问题还是色谱柱的原因?不胜感激!
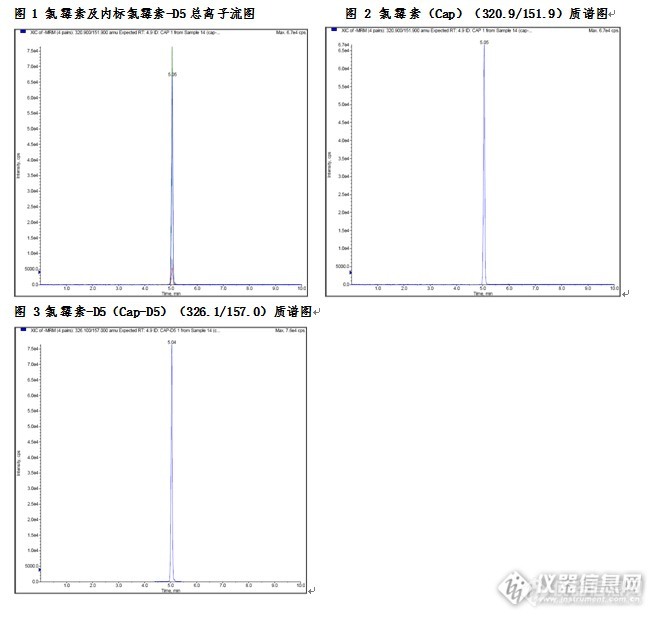
动物源性食品中氯霉素检测的固相萃取方法一、实验目的本研究利用固相萃取法作为样品的前处理方法,LC-MS法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂的使用,操作简便。 二、实验目标物氯霉素(CAS:56-75-7) 三、应用范围本方法适用于动物源性食品中氯霉素的LC-MS/MS检测及确证。 四、参考标准推荐性国家标准《GB/T 22338-2008动物源性食品中氯霉素类药物残留量测定》 五、实验材料 Biocomma®Silibase™ C18固相萃取柱6mL/1000mg。六、实验方法 1、样品提取 将称取5g试样,精确到0.01g。置于50ml于涡旋具盖离心管中,加入5ml水,于涡旋混合器上快速混合1min,使试样完全溶解。准确加入15ml乙酸乙酯,在振荡器上振荡10min,以3000r/min离心10min,准确吸取上层乙酸乙酯12ml转入15ml的离心管中,置于浓缩吹氮仪在45℃吹扫蒸干,加入5ml水溶解,待净化。 2、SPE柱净化(1)活化:依次采用5mL甲醇、5mL水活化C18固相萃取小柱。(2)净化:将提取液过C18固相萃取柱,待溶液完全流出后,用2×3ml水过柱,然后再用5ml乙腈+水淋洗柱,弃去全部淋出液。用真空泵减压抽干10min。 3、洗脱 用6ml乙酸乙酯洗脱,收集洗脱液于10ml离心管中,于45℃用氮气吹干液吹干,用乙腈+水(2:8,体积比)定容至1ml,供LC-MS/MS上机测定。 4、LC-MS条件色谱柱:Venusil ASBC18(2.1×150mm,5µm,100Å);质谱仪:API 4000流动相:A:0.1mM乙酸铵溶液(0.1%甲酸) B:甲醇溶液;流速:0.2mL/min柱温:40℃进样体积:5ul离子源:电喷雾(ESI),负离子模式检测方式:多反应监测(MRM)。 表1 质谱仪离子源参数 Source/Gas Collision Gas(CAD) 6 Curtain Gas(CUR) 30 Ion Source Gas 1(GS 1) 50 Ion Source Gas 2(GS 2) 50 Ion Spray Voltage(IS) -4500 Temperature(TEM) 550 Interface Heater(ihe) On 表2 氯霉素及内标的母离子和子离子参数表 物质名称 保留时间(min) 监测离子对 DP EP CE CXP 氯霉素 4.90 320.9/151.9 -60 -10 -25 -12 320.9/256.9 -60 -10 -25 -12 氯霉素-D5 4.90 326.1/157.0 -60 -10 -25 -12 七、实验结果1、10ppb猪肉基质氯霉素添加回收结果平均加标回收率在90%以上,RSD值小于5符合标准要求。 表3 猪肉基质氯霉素添加回收结果 名称 1(%) 2(%) 3(%) 平均回收率(%) RSD(%) 氯霉素 98.31 96.28 90.61 95.07 4.20 2、 空白样品添加氯霉素残留物色谱图http://ng1.17img.cn/bbsfiles/images/2015/08/201508141639_560777_3310_3.jpg
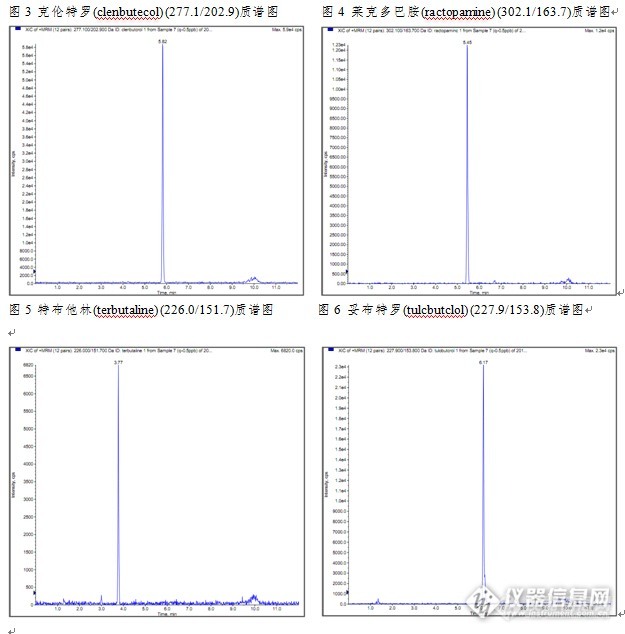
动物源性食品中β-受体激动剂检测的固相萃取方法一、实验目的本研究利用固相萃取法作为猪肉样品的前处理方法,LC-MS/MS法作为检测手段。该方法可简化猪肉样品的前处理过程,节省有机溶剂的使用,操作简便。 二、实验目标物五种β-受体激动剂物质:克仑特罗(CAS:37148-27-9),沙丁胺醇(CAS:18559-94-9),莱克多巴胺(CAS:97825-25-7),特布他林(CAS:23031-25-6),妥布特罗(CAS:41570-61-0)。 三、应用范围本方法适用于动物源性食品中β-受体激动剂LC-MS/MS检测及确证。 四、参考标准推荐性国家标准《GB/T 21313-2007 动物源性食品中β-受体激动剂残留检测方法液相色谱-质谱/质谱法》五、实验材料MCX固相萃取柱6mL/200mg。六、实验方法 1、样品提取 称取均质试样10.0g试样(精确至0.01g),加入15mLpH 5.2的乙酸-乙酸铵缓冲溶液,1000 r/min 匀浆1 min,再加入β-噗糖醛酸苷肽酶/芳香磺酸酯酶溶液100 μL,于37℃±1℃振荡酶解过夜。取出冷却后,10℃9500 r/min离心10 min。取全部上清液,待净化。 2、SPE柱净化(1)活化:加入6mL5%甲醇氨、6mL甲醇、6mL水、6mL 0.1mmol/L高氯酸(pH 4.0)溶液活化。(2)上样和洗脱:当溶剂液面到达柱吸附层表面时,立即倒入上述待净化溶液,过柱,弃去滤液,用2mL甲醇、2mL 2%甲醇水溶液淋洗,最后用7mL5%甲醇氨洗脱。(3)重新溶解:洗脱液缓慢氮气流条件下吹至近干,用甲醇-0.1%甲酸水溶液溶解定容至1.0mL,涡旋混匀1min,用于LC-MS/MS测定。 4、LC-MS条件色谱柱:Venusil ASB C18(2.1×150mm,5µm,100Å);质谱仪:API 4000流动相:A:10mM乙酸铵(0.1%甲酸)溶液 B:甲醇溶液;表1梯度洗脱条件 时间/min B/% A/% 0.00 5 95 4.00 50 50 6.00 10 90 8.00 95 5 8.01 5 95 12.00 Stop 流速:0.2mL/min 柱温:40℃ 进样体积:2μL 离子源:电喷雾(ESI),正离子模式 检测方式:多反应监测(MRM)表2质谱仪离子源参数 Source/Gas Collision Gas(CAD) 10 Curtain Gas(CUR) 30 Ion Source Gas 1(GS 1) 50 Ion Source Gas 2(GS 2) 50 Ion Spray Voltage(IS) 5500 Temperature(TEM) 550 Interface Heater(ihe) On 表3β-受体激动剂的母离子和子离子参数表 物质名称 保留时间(min) 监测离子对 DP EP CE [align
2013年10月25日,在哈尔滨邮区中心局进境邮件查验现场,一个来自日本广岛大学,寄往本市某科研单位、申报为抗体的邮件引起了检验检疫人员的警惕。经对收件人电话问询,在没有确定邮寄物来源及成份前,无法认定该批货物的风险等级,随即进行了开箱查验。开箱后发现11个小指型试剂管和2只冰袋,初步认定为科研用、兔源性、细胞标识用单克隆抗体。 鉴于该批货物未随附任何证明材料,为保障进境动物源性生物材料及制品的质量安全,防止境外动物疫病疫情传入,检验检疫人员根据相关规定,对该批货物暂作截留处理,并出具了《检验检疫处理通知书》,由邮局转交收件人。这是哈尔滨邮局办事处首次截获的动物源性生物制品。 检验检疫部门在此提醒相关教学科研及个人,自觉遵守国家相关法律法规,在邮寄动植物、动植物产品及特殊物品时,提前到检验检疫部门咨询,办理相应的检疫审批手续及证明,以免给国家和个人带来损失。一般邮寄生物制品的时候,需要办什么手续呢?你们都是怎么做的呢?
日前,欧盟食品安全局(EFSA)就其转基因动物源性食品和饲料的风险评估(包括动物健康和福利方面)指南草案,展开公众评议;评议截止日期为2011年9月30日。在该指南草案中,EFSA就申请转基因动物源性食品和饲料欧盟上市许可所须风险评估的具体数据要求和研究方法做出了规定。该风险评估方法是将转基因动物源性食品和饲料与常规动物源性产品进行比对;评估包括食品/饲料安全和动物健康及福利等方面内容。当前的指南草案规定的风险评估方法假定传统饲养动物源性食品和饲料具有长期使用安全史,从而将其作为转基因动物源性食品和饲料的风险评估基准。指南规定了开展风险评估对比分析所需的具体数据要求。风险评估的重要内容包括分子特征,成分分析和毒性、营养及潜在过敏性评估。草案还列明了转基因动物健康和福利对比评估的方法。该评估主要用于两个方面:一是对转基因动物自身,评估重点应放在动物身体系统的正常功能(比如抗病能力);二是对食品和饲料风险评估,因为动物的健康和福利状况被视为动物源性产品安全性的一个重要指标。草案强调了广泛开展转基因动物与传统动物特征和特性(包括生理参数)对比分析的必要性。草案还建议应在转基因动物生长的所有阶段进行动物健康和福利评估,直至其要获得授权的时点。草案建议的三阶段评估策略包括转基因动物最初生长的实验室环境,实验室外大量动物的实地试验以及在商业环境下的大量动物试验(授权前)。草案的最后还针对转基因动物和转基因动物源性食品和饲料的上市后监督(PMM)提出了建议,以便在相关产品获准上市后,发现其可能出现的与转基因相关的潜在不良影响。针对食品和饲料的安全问题,PMM是对全面上市前毒性测试计划的有力补充。针对一些特定情况,如营养成分改变的转基因食品和饲料,以及/或改变基因以实现特定健康效果的食品和饲料,均应实施PMM。针对动物健康和福利,PMM则旨在发现可能存在的发生率较低的长期不良性影响。
建立测定动物源性食品中金刚烷胺的固相萃取/液相色谱-电喷雾串联质谱分析方法。以甲醇-1%三氯乙酸(50+50,v/v)作提取溶剂,采用超声波辅助溶剂萃取法萃取动物源性食品,萃取液用Waters Oasis MCX 固相萃取柱进行净化浓缩。以Thermo Hypersil Gold C18色谱柱为分离柱,在正离子模式下以电喷雾电离串联质谱仪进行测定。对流动相组分和流动相添加剂对质谱的离子化效率进行考察,在1.0~50.0?ng/mL范围内线性关系良好(r?≥?0.99)。样品在5.0μg/k,10.0μg/k和20.0μg/kg添加水平的回收率为75.9%~108.5%,相对标准偏差小于8.0%;方法的检出限为5.0μg/kg。本方法具有很高的灵敏度和准确度,能够满足动物源性食品中的金刚烷胺残留量的快速、高灵敏检测分析。
动物源性食品己烯雌酚检验方法—高效液相色谱法1 范围本方法规定了动物源性食品中己烯雌酚残留量的快速测定方法。原理试样中的残留的己烯雌酚经快速检测前处理试剂盒提供的试剂提取、浓缩、净化后用液相色谱进行检测,外标法定量。3 试剂和材料除另有规定外,所有试剂均为分析纯,水为重蒸馏水。3.1乙腈:色谱纯。3.2冰乙酸:色谱纯。3.3甲醇:色谱纯。3.4无水乙酸钠。3.5对-甲苯磺酸。3.6乙酸盐缓冲液:称取4.950g无水乙酸钠及0.950g对-甲苯磺酸,溶解于950mL水中,用冰乙酸调节溶液pH到4.5,最后用水定溶到1000mL。3.7标准品: 己烯雌酚3.8标准贮备溶液:分别称取标准品各10.0mg,用甲醇溶解后转移至10mL棕色容量瓶中,用甲醇定容,于—20℃条件下保存。使用时,用甲醇稀释上述标准储备溶液,配制成不同浓度的标准工作液。3.9己烯雌酚快速检测前处理试剂盒*。4 仪器和设备4.1高效液相色谱仪:配紫外-可见检测器。4.2匀浆机。4.3旋转蒸发仪。4.4离心机:4 000 r/min4.5聚四氟乙烯离心管:100 mL,具塞。4.6微孔滤膜:0.45 µ m。5测定步骤5.1提取、浓缩、净化准确称取已捣碎的样品10.00 g于50 mL离心管中,先加己烯雌酚快速检测前处理试剂盒中的提取剂(液体20.0 mL)、8000r/min均质1min,于4000 r/min离心5 min,取出上清液10mL用水稀释至30mL。残留物用2.0 mL蒸馏水溶解。取稀释液于层析柱中(使用前依次用5mL甲醇、5mL蒸馏水激活)挤干,加5mL洗涤剂洗涤挤干,加5mL洗脱剂洗脱,接收洗脱液,50℃水浴旋转蒸干,用1mL甲醇溶解残渣于0.45 µ m微孔滤膜过滤,供仪器测定。5.2绘制标准工作曲线移取己烯雌酚混和标准溶液, 50µ g/kg、100µ g/kg、200µ g/kg、500µ g/kg、1000µ g/kg标准工作溶液,样液用高效液相色谱仪测定,得出标准工作曲线。5.3测定5.3.1液相色谱条件a) 色谱柱: C18柱,150 mm×4.6 mm(i.d.),粒度5m b) 流动相:乙腈+乙酸盐缓冲液 (70+20,体积分数) c) 流速:1.0 mL/min d) 柱温:室温 e) 检测波长:250nmf) 进样量:20 L。5.3.2色谱测定根据样液中己烯雌酚的含量情况,选定峰面积相近的标准工作溶液。标准工作溶液和样液中的己烯雌酚响应值均应在仪器的检测线性范围内。5.3.3空白实验除不加试样外,按上述测定步骤进行。5.3.4结果计算用色谱数据处理机或按式(1)分别计算供试样品中的己烯雌酚残留量。 2 ci×Vω= …… (1)mω-样品中己烯雌酚残留量,μg/kg;ci -标准曲线上查出试样溶液中己烯雌酚标准工作溶液的浓度,μg/L;2-换算常数;V-最终定容体积数,mL;m-供试试料样品重量,g。6测定低限、回收率6.1检测限本方法的检测限己烯雌酚为10μg/kg。6.2回收率本方法回收率己烯雌酚为:85%~105%。
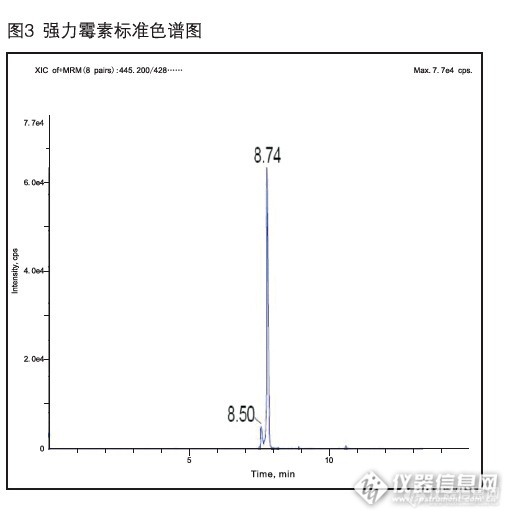
动物源性食品中四环素类药物检测的固相萃取方法(C8/SCX固相萃取柱)一、实验目的本实验利用固相萃取法作为样品的前处理方法,LC-MS/MS法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂用量。 二、实验目标物四环素(CAS:60-54-8),金霉素(CAS:57-62-5),强力霉素(CAS:564-25-0)。三、应用范围本方法适用于动物源性食品中四环素类药物的LC-MS/MS检测及确证。四、参考文献 推荐性国家标准《GB/T 21317-2007 动物源性食品中四环素类兽药残留量检测方法 液相色谱-质谱/质谱法与高效液相色谱法》。 五、实验材料 C8/SCX固相萃取柱200mg/6mL。 六、实验方法 1、样品提取 将样品混匀、均质,有结晶的样品于60℃以下水浴融化混匀、迅速冷却;在50 mL带螺盖的离心管中精确称取约6g(精确到0.01g)均质后的样品;加入25 mLEDTA-Mcllvaine缓冲溶液迅速混匀1min,冰浴超声20 min;3000 r/min离心10min,取上清液,过快速滤纸,收集滤液,待净化。 2、SPE柱纯化 (1)活化:将固相萃取真空装置与真空泵连接好,装上C8/SCX柱,依次以5mL甲醇和10 mL纯水预处理。 (2)上样和洗脱:取样品提取液中上清液10mL过柱,流速控制在约每秒1滴;依次以5mL水+5mL甲醇/水(1:19,体积比)溶液淋洗柱子;淋洗液完全通过小柱后,抽真空20min;以10 mL甲醇+乙酸乙酯(1+9)洗脱,收集洗脱液。 (3)浓缩定容:将洗脱液氮吹吹干,用2mL甲醇定容,过0.22μm有机滤膜,用于LC-MS/MS分析。 5、LC-MS/MS条件 液相色谱-质谱/质谱仪 色谱柱:C18柱:150mm×2.1mm,2.0μm,或相当者 流动相:甲醇-10mM草酸七、实验结果1、添加回收结果 向样品中加入不同水平的四环素类药物,回收率结果如下:(见表1)表1 动物组织中四环素类药物添加回收结果 样品名称 化合物名称 添加水平(ng/mL) 回收率(%) 猪肉 四环素 50 97.38 100 92.94 强力霉素 50 80.43 100 81.87 金霉素 50 95.72 100 94.63 2、 空白样品添加农药残留物色谱图http://ng1.17img.cn/bbsfiles/images/2015/08/201508141645_560791_3310_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508141645_560792_3310_3.jpg
定动物源性食品中11种_2_受体激动剂的研究[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=122366]定动物源性食品中11种_2_受体激动剂的研究[/url]
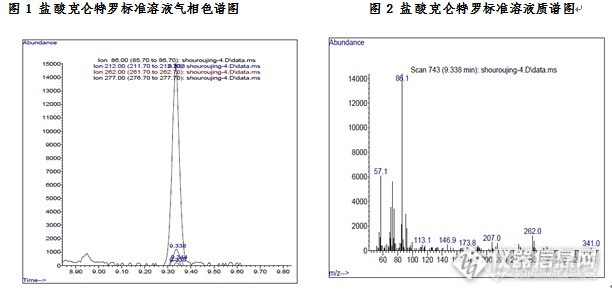
动物源性食品中盐酸克仑特罗检测的固相萃取方法(Silibase™ C8/SCX)一、实验目的本研究利用固相萃取法作为猪肉样品的前处理方法,GC-MS法作为检测手段。该方法可简化猪肉样品的前处理过程,节省有机溶剂的使用,操作简便。 二、实验目标物盐酸克仑特罗(CAS;21898-19-1)三、应用范围本方法适用于动物源性食品中盐酸克仑特罗的GC-MS检测及确证。 四、参考标准农业部推行标准《NY/T 468-2006 动物组织中盐酸克伦特罗的测定气相色谱-质谱法》五、实验材料 C8/SCX固相萃取柱6mL/500mg。六、实验方法1、样品提取 称取均质试样5.0g试样(精确至0.01g)于50mL离心管中,加入15mL乙酸乙酯,再加入3mL10.0%碳酸钠溶液,然后以10000r/min以上的速度均质60s。盖上盖子以5000r/min的速度离心2min,吸取上层有机试剂于离心管中,在残渣中再加入10mL乙酸乙酯在旋涡混合器上混合1min,离心后吸取有机溶剂并合并提取液。在收集的有机试剂中加入5mL 0.1mol/L的盐酸溶液,涡旋混合30s。以5000r/min的速度离心2min,吸取下层溶液,同样步骤重复萃取一次,合并两次萃取液,用2.5mol/L氢氧化钠溶液调节pH至5.2。 2、SPE柱净化(1)活化:向C8/MCX复合柱小柱中依次加入5mL甲醇、水5mL和5mL 30mmol/L盐酸润洗固相萃取小柱。(2)上样和洗脱:将上述备用液过柱,依次用5mL水、5mL甲醇淋洗,真空抽干,用4%氨化甲醇5mL洗脱小柱,收集洗脱液。(3)衍生化及检测:0℃缓慢氮气流条件下吹至近干,加入甲苯100μL和双三甲基硅基三氟乙酰胺100μL,涡旋震荡20s,密封玻璃塞,置于80℃恒温烘箱中加热1h,冷却后加300μL甲苯,作为试样溶液,供气相色谱-质谱分析,上气相色谱-质谱仪测定。3、GC-MS条件气相色谱-质谱仪;色谱柱:HP-5MS进样口:220℃柱温:70℃(保持0.6min),以25℃/min升温至200℃(保持6min),以25℃/min升温至280℃(保持5min)载气:高纯He,流速:0.9mL/min进样体积:1μlGC/MS传输线温度:280℃溶剂延迟:8min分析器温度:230℃四级杆温度:150℃ 七、实验结果1、添加回收结果实验结果表明,C8/SCX复合固相萃取柱适用于动物组织中盐酸克伦特罗的预处理,能净化动物组织样品,实验加标回收率及RSD能满足定量实验的要求。表1动物源性食品中盐酸克伦特罗的添加回收结果 1 2 3 4 5 平均回收率(%) RSD(%) n=5 回收率(%) 85.6 92.4 94.1 95.7 87.6 90.8 4.23 2、标准溶液色谱图在GC-MS操作条件下,得到标准溶液色谱图如图1和图2http://ng1.17img.cn/bbsfiles/images/2015/08/201508141658_560811_3310_3.jpg
动物源性食品中兽药残留的检测兽药残留检测项目针对性比较强,往往只会检测某一种或某一类药物,这比较适合采用“保留目标化合物模式”进行净化,具有净化效果好、回收率高的特点。下表是选择SPE柱的指导办法:目标化合物类型推荐中性、弱酸和弱碱性药物比如氯霉素、磺胺、呋喃类代谢物等ProElut PLS亲水亲脂平衡柱酸性药物(含有羧基或酚羟基)比如沙星类药物ProElut PXA混合型阴离子交换反相柱碱性药物(含有氨基)比如磺胺ProElut PXC混合型阳离子交换反相柱强阴离子药物(含有磺酸基、磷酸基)ProElut PWA混合型弱阴离子交换反相柱强阳离子药物(含有季铵基)ProElut PWC混合型弱阳离子交换反相柱
附件:中华人民共和国2006年度进出口动物及动物源性食品残留物质监控抽样及检测计划(2006. 1-2006.12)中华人民共和国国家质量监督检验检疫总局 目 录●起草说明●附录1 2006年对出口动物源性食品监控的残留物质与动物种类表●附录2 2006出口动物及动物源性食品残留物质监控抽样计划表●附录3 2006年对进口动物源性食品监控的残留物质与动物种类表●附录4 2006进口动物及动物源性食品残留物质监控抽样计划表●附录5-1 2006年出口残留监控计划各局抽样与检测样品数目统计表●附录5-2 2006年进口残留监控计划各局抽样与检测样品数目统计表●附录5-3 2006年进出口残留监控计划各局抽样与检测样品数目统计表●附录6 残留监控的取样流程图和有关单证●附录7 基准实验室及其分工●附录8 残留监控样品接受人通讯录●附录9 残留监控标准物质供应中心通讯录2006年度进出口动物及动物源性食品残留监控抽样及检测计划起草说明为提高我国动物源性食品的安全卫生质量,保护人民的身体健康,增强我国动物源食品在国际市场上的竞争力,结合检验检疫的出口业务需要和实际检测能力,在总结过去5年半残留监控工作经验的基础上,重点考虑2005年监控检出和阳性情况以及出口国对我国产品的预警通报情况,同时兼顾其他国家对动物及动物源性食品的安全卫生要求,提出了《中华人民共和国2006年度出口动物及动物源性食品残留物质监控抽样及检测计划》、《中华人民共和国2006年度进口动物及动物源性食品残留物质监控抽样及检测计划》。现对有关修订情况说明如下:一、 实施时间2006年度残留监控计划的实施时间为2006年1月至12月。二、残留监控计划修订的依据和原则(一)针对我国在2005年残留监控计划实施过程中的情况和问题,如产品的调整或进出口数量的变化,适当调整各地区的取样数。(二)根据上年度残留监控结果反馈情况,特别是阳性结果的检出情况,同时根据各国预警通报的情况,适当增加对应动物和检测项目的取样数量;对于历年监测均为阴性的项目, 适当减少对应动物的取样数量。总体取样量比去年略有上升。(三) 在对过去2年进口产品监控结果数据分析的基础上,在2006年对各个项目的抽样量进行调整。针对出口情况的变化,取消了个别动物品种的监控。同时也对检测的药物种类进行了调整,取消了个别项目。(四)根据日本肯定列表的情况和我国实际检测能力,适当增加监测项目。三、主要修订之处(一)在《中华人民共和国2006年度出口动物及动物源食品残留物质监控抽样及检测计划》中:1.监控的物种、样品基质及残留标识物。(1)鉴于2005年马产品无出口,无法取样,故取消对马组织的残留监控。(2)对于牛的监控,左旋咪唑、阿维菌素、伊维菌素的监测样品基质由牛肾调整为牛肝(参照欧盟标准)。(3)对于鸡的监控,左旋咪唑、阿维菌素的监测样品基质由鸡肾调整为鸡肝(参照欧盟和日本标准)。(4)对于氯霉素残留的监控,检测样品基质由肝组织调整为肌肉组织。(5)对于硝基咪唑类残留的监控,检测样品基质由肝组织调整为肾组织,并明确需要同时检测其代谢产物2-羟甲基-1-甲基-5-硝基咪唑。(6)对于链霉素的监控,明确需同时检测其代谢产物双氢链霉素。(7)对于多氯联苯的监控,检测样品基质调整为脂肪组织。(8)对于苯并咪唑的监控,明确需同时检测其代谢产物。2.增加的检测项目。(1)牛:增加甲基氢化泼尼松、氟苯尼考、麻保沙星、萘夫西林、磺胺氯哒嗪、磺胺胍、磺胺甲氧哒嗪、氧化丙硫咪唑、氟苯哒唑、甲苯咪唑的监控。(2)羊:增加萘夫西林、磺胺氯哒嗪、磺胺胍、磺胺甲氧哒嗪、氧化丙硫咪唑、氟苯哒唑、甲苯咪唑的监控。(3)猪:增加莱克多巴胺、萘夫西林、甲砜霉素、磺胺氯哒嗪、磺胺胍、磺胺甲氧哒嗪、丙硫哒唑、氧化丙硫咪唑、氟苯哒唑、甲苯咪唑的监控。(4)禽:增加甲基氢化泼尼松、氟苯尼考、马杜霉素、萘夫西林、磺胺氯哒嗪、磺胺胍、磺胺甲氧哒嗪、丙硫哒唑、氧化丙硫咪唑、氟苯哒唑、甲苯咪唑的监控。(5)兔:增加甲基氢化泼尼松、氟苯尼考的监控。(6)水产品:增加磺胺氯哒嗪、磺胺胍、磺胺甲氧哒嗪、结晶紫的监控。(7)蛋:增加氟苯尼考的监控。3.调整的检测限和最高残留限量。(1)牛肝:磺胺喹恶啉检测限由20μg/kg调整50μg/kg,最高残留限量由20μg/kg调整为100μg/kg(参照日本标准)。(2)猪肝:强力霉素最高残留限量由300μg/kg 调整为50μg/kg(参照日本标准);伊维菌素最高残留限量由15μg/kg调整为100μg/kg (参照欧盟标准);磺胺喹恶啉检测限由20μg/kg调整为50μg/kg,最高残留限量由20μg/kg调整为100μg/kg(参照日本标准)。(3)鸡肝:强力霉素最高残留限量由300μg/kg 调整为50μg/kg(参照日本标准);链霉素的检测限100μg/kg 调整为10μg/kg,最高残留限量由500μg/kg 调整为 10μg/kg(参照日本标准);磺胺二甲嘧啶检测限由20μg/kg调整为50μg/kg,最高残留限量由20μg/kg调整为100μg/kg(参照日本标准);盐霉素最高残留限量由1800μg/kg调整为500μg/kg(参照日本标准);莫能菌素最高残留限量由50μg/kg调整为500μg/kg (参照日本标准);恩诺沙星、环丙沙星最高残留限量由200μg/kg调整为10μg/kg ,两者之和小于200μg/kg调整为小于10μg/kg(参照日本标准);沙拉沙星最高残留限量由100μg/kg调整为80μg/kg(参照日本标准); 左旋咪唑最高残留限量由10μg/kg调整为100μg/kg(参照日本标准);鸡肾的尼卡巴嗪检测限由10μg/kg调整为50μg/kg(参照日本标准);氯氰菊酯最高残留限量由200μg/kg调整为50μg/kg(参照日本标准)。(4)兔肝:甲砜霉素最高残留限量由50μg/kg调整为未制定。(5)鳗鱼:恩诺沙星、环丙沙星、诺氟沙星、氧氟沙星检测限由20μg/kg调整为10μg/kg,最高残留限量由ND调整为100μg/kg、100μg/kg、10μg/kg、100μg/kg,恩诺沙星与环丙沙星残留之和不得超过100μg/kg(参照欧盟和日本标准);磺胺类检测限由20μg/kg调整为10μg/kg(磺胺二甲嘧啶未改变),最高残留限量由ND调整为10μg/kg(磺胺二甲氧嘧啶调整为100μg/kg)(参照日本标准);(6)养殖虾:磺胺二甲嘧啶检测限由10μg/kg调整为20μg/kg;恶喹酸最高残留限量由50μg/kg调整为100μg/kg(参照日本标准)。(7)蜂王浆:链霉素最高残留限量由20μg/kg调整为未制定。(8)蜂蜜:链霉素最高残留限量由20μg/kg调整为未制定。(二)在《中华人民共和国2005年度进口动物及动物源食品残留物质监控抽样及检测计划》中:1.增加的检测项目及其检测限与最高残留限量。(1)禽类:增加甲砜霉素、磺胺氯哒嗪、磺胺胍、磺胺甲氧哒嗪、克球酚的监控。(2)水产品:增加氟苯尼考、甲砜霉素的监控。(3)奶制品:增加甲砜霉素、苄青霉素、邻氯青霉素、双氯青霉素、亚硝酸盐的监控。(4)牛组织:增加氟苯尼考、甲砜霉素的监控。(5)猪组织:增加甲砜霉素、莱克多巴胺、二恶英的监控。各监控产品增加的监控项目的检测限和最高残留限量详见附录。2.物种和抽样地区变化情况。(1)对水产品的监控:增加对云南、广西、宁波局辖区的监控,减少福建局抽样量。(2)对牛的监控:去掉深圳、山东局的抽样,减少天津局抽样量。(3)对甲壳类的监控:去掉深圳局,新增宁波局,增加上海局抽样量,减少辽宁局抽样量。(4)奶粉的监控:新增湖南局辖区的监控,减少上海、辽宁局抽样量,增加深圳局抽样量。(5)对猪组织的监控:减少山东局抽样量。(6)对禽类的监控:减少天津局抽样量。(7)对肠衣的监控:减少江苏、天津局的抽样数。四、计划外残留监控对鹅、野禽等禽类产品,牛娃,贝类、蟹、海蛰、鱿鱼等水产品的残留监控,2006年仍不列入统一的残留监控计划。请各有关直属局根据出口业务的需要并结合本局的检测能力,分别参照鸡或鱼的残留监控项目开展残留监控工作;监控结果按照有关《残留监控结果统计报表》的规定上报国家质检总局。
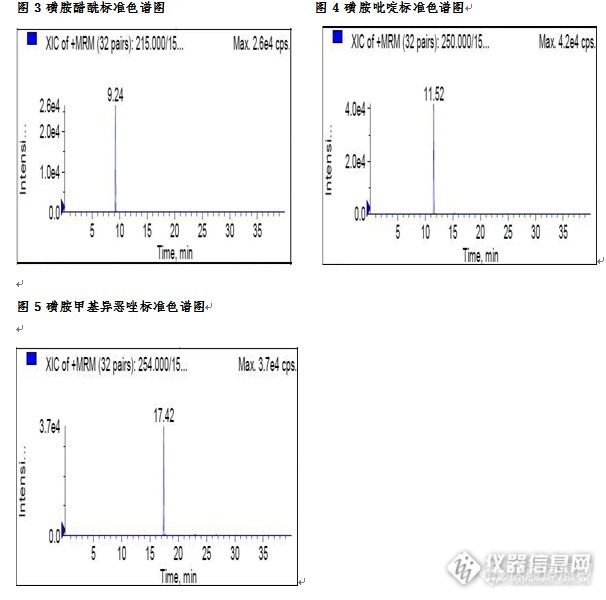
动物源性食品中磺胺类药物检测的固相萃取方法(Silibase™ ALN)一、实验目的本实验利用固相萃取法作为样品的前处理方法,LC-MS/MS法作为检测手段。该方法可简化样品的前处理过程,节省有机溶剂用量。 二、实验目标物磺胺噻唑(CAS:72-14-0),磺胺甲基嘧啶(CAS:127-79-7),磺胺醋酰(CAS:144-80-9),磺胺吡啶(CAS:144-83-2),磺胺甲基异恶唑(CAS:127-69-5)。三、应用范围本方法适用于动物源性食品中磺胺类药物的LC-MS/MS检测及确证。四、参考文献 推荐性国家标准《GB/T 21316-2007 动物源性食品中磺胺类药物残留量的测定 液相色谱-质谱/质谱法》。 五、实验材料 Biocomma®Silibase™ ALN固相萃取柱500mg/3mL。 六、实验方法 1、样品提取 将称取5g 试样,精确到0.01g。置于50mL涡旋具盖离心管中,加入5mL水,于涡旋混合器上快速混合1min,使试样完全溶解。准确加入15mL乙酸乙酯,在振荡器上振荡10min,以3000r/min 离心10min,准确吸取上层乙酸乙酯12mL转入15mL的离心管中,置于浓缩吹氮仪在45℃吹扫蒸干,加入5mL水溶解,待净化。 2、SPE净化 将提取液过中性氧化铝柱,待溶液完全流出后,用2×3mL水洗柱,然后再用5mL乙腈+水淋洗柱,弃去全部淋出液。用真空泵减压抽干10min,最后用6mL乙酸乙酯洗脱,收集洗脱液10mL离心管中,于45℃用氮气吹干液吹干,用乙腈+水(20+80)定容至1mL,供LC-MS/MS 上机测定。 3、LC-MS/MS条件 液相色谱-质谱/质谱仪 色谱柱:C18柱:150mm×2.1mm,2.0μm,或相当者 流动相:乙腈-0.1%甲酸七、实验结果1、添加回收结果 向样品中加入不同水平的四环素类药物,回收率结果如下:(见表1)表1 动物组织中四环素类药物添加回收结果 样品名称 化合物名称 添加水平(ng/mL) 回收率(%) 猪肉 磺胺噻唑 50 84.76 100 89.44 磺胺甲基嘧啶 50 81.30 100 91.08 磺胺醋酰 50 92.92 100 94.37 磺胺吡啶 50 81.74 100 88.09 磺胺甲基异恶唑 50 81.81 100 88.22 2、 空白样品添加农药残留物色谱图http://ng1.17img.cn/bbsfiles/images/2015/08/201508141631_560757_3310_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/08/201508141631_560758_3310_3.jpg
内部培训资料:粮食、果蔬、动物源食品样品取样部分要求,根据GB 2763-2021中附录A编制,有需要的拿去[img]https://simg.instrument.com.cn/bbs/images/default/em09502.gif[/img]
动物源性食品中四环素类兽药残留量检测方法,供各位版友参考!!!
【关键词】 动物源食品 磺胺类药物残留 GPC法测定磺胺类药物是一类广泛使用的预防性和刺激动物生长(常作饲料添加剂)的抗生素,其在动物源性食品中的残留对人类健康的潜在危害日趋严重,被受到广泛关注。进行动物源性食品磺胺类残留分析具有重要意义〔1〕。由于动物源食品基质复杂,在进行残留分析时通常需要对样品提取液进行净化,目前报道的净化技术主要有液液萃取、固相萃取、基质分散固相萃取〔2-5〕等方法。凝胶渗透色谱(GPC)方法作为一种通用的样品净化技术已经成功应用于农药的多残留分析〔6〕,但应用于药物的多残留分析报道很少〔7〕。为了探讨GPC净化技术应用于动物源食品中药物残留分析的效果,本研究应用GPC净化技术结合高效液相色谱法(HPLC)检测了动物源食品中9种磺胺类药物残留量,取得较为满意的结果。现报告如下。
CAC/GL 68-2008 Guideline for the Conduct of Food Safety Assessment of Foods Derived from Recombinant-DNA Animals中文名称:动物源性DNA重组食品安全评估指导方针[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=166131]CAC/GL 68-2008 动物源性DNA重组食品安全评估指导方针(英文)[/url]
[color=#333333]请问为什么要对动物源性食品中氟喹诺酮类药物残留进行检测[/color][color=#333333] [/color]
本方法适用于动物源性食品中14种喹诺酮药物残留的检测,供大家参考!!!
8月24日,澳大利亚检验检疫局(AQIS)发布公共检疫警告(PQA0745)称,在与旅客和邮包计划处磋商后,AQIS生物进口计划处制定了除用作动物性食品、肥料或饲育目的之外的动物肠源性产品(不包括肠衣)新进口条件。新进口条件适用于羊肠线材料、乐器弦和动物源性网球拍线,具体内容如下:一、非商业性进口条件1. 个人、文化演出团体或运动队携带入境(不论随身携带或邮寄)的供自己使用的乐器或运动器材上附着的动物源性弦/线,不需要进口许可证和入境检疫。2. 不符合上述要求的货物适用商业性进口条件。二、商业性进口条件1. 需要进口许可证,并且须在货物进口到澳大利亚时有效。进口许可证申请须送至AQIS在堪培拉的办事处进行评估。2. 每批货物须接受入境检疫。3. 进口许可证条件要求货物须随附关于产品成分和相关加工方法的生产商声明。根据与成分相关的检疫风险水平,可能还需要官方证书。4. 当货物未随附有效文件或随附带错误声明的文件时,将被要求修正文件。5. 进口许可证条件要求货物到达时需接受检验,以确保未受到外来杂质的污染及/或侵染。如发现污染及/或侵染,将按照AQIS认可的适当方法对外来杂质进行处理。6. 整箱集装箱内的木质包装、木托盘或垫木在到达时也需接受检验和处理,除非证明已按照AQIS认可的方法进行过处理。此外,该进口条件还规定了提交给AQIS的所有文件须符合最低文件要求政策和非商品信息要求政策,以及进口许可证费用。该进口条件的详细内容见以下网址:http://www.aqis.gov.au/icon32/asp/ex_casecontent.asp?intNodeId=8941522&intCommodityId=27866&Types=none&WhichQuery=Go+to+full+text&intSearch=1&LogSessionID=0
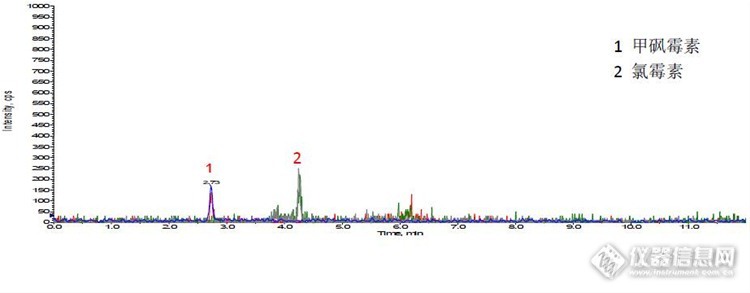
喹诺酮是一类合成抗生素类药物,治疗系统感染疾病,并且在动物饲养中作为预防和治疗药物普遍使用。近年来,这些药物在动物组织中的残留已被引起广泛关注。联合国粮农组织、世界卫生组织食品添加剂专家联合委员会、欧盟都已制定了多种喹诺酮类药物在动物组织中的最高残留限量。 喹诺酮类药物各种残留分析方法,主要包括高效液相法(HPLC)以及与此相关的HPLC-UV、HPLC-FD、HPLC-DVD、LC-MS/MS、LC-ESI-MS/MS,另外还有荧光光谱法、毛细管电泳法和酶联免疫法等。样品前处理主要采用SPE、GPC、液液萃取等净化方法;但是目前还没有文献采用QuEchERS方法净化。而且官方所发布标准的检测方法中大多数是LCMS-SPE或者LCMS-LLE。 本方法基于QuEchERS方法原理,采用乙腈提取,然后取出1 mL净化,上机分析。本方法前处理过程简单、方便,并能同时检测马波沙星、恩诺沙星、沙拉沙星、洛美沙星、双氟沙星、萘啶酸、氟甲喹;节省时间,降低基质效应。回收率达85%以上,保证实验结果的准确性、重现性;本方案方法检出限均为1.0 ug/kg,优于国标方法《GB/T 21312-2007 动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱 质谱法》,可供广大分析工作者使用。1、适用范围 适用于草鱼、猪肉、牛奶、蜂蜜等动物源性食品中氯霉素、甲砜霉素等药物残留量的测定。本方法检出限0.1ug/kg。2、提取(1) 猪肉、草鱼样品取5.0 g样品与2.0 g氯化钠,搅匀,加5 mL乙腈,涡旋混合1 min,振荡5 min,6000rpm下离心2 min,取 1mL上清液待净化。(2) 牛奶样品取5.0 g样品与2.5 mL乙腈,涡旋混合1 min,加2.0 g氯化钠和2.5 mL乙腈,涡旋混合1 min,6000 rpm下离心2 min,取 1mL上清液待净化。(3) 蜂蜜样品取5.0 g样品,加5 mL水溶解,涡旋1 min,加2.0 g氯化钠和5 mL乙腈,涡旋混合1 min,6000 rpm下离心2 min,取 1mL上清液待净化。 3、净化将1 mL 提取液转移到ProElut QuE 2mL Tube(Cat#:64531),涡旋混合30 S,10000 rpm下离心1 min,取上清液500 μL于浓缩管中,加水100 μL,涡旋混匀,室温下氮吹至剩余约100 μL溶液,加水定容至500 μL,混匀,过0.22 μm微孔滤膜,进LC-MS/MS分析。 4、分析条件4.1 UPLC 条件:色谱柱:Endeavosil C18,100×2.1 mm,1.8 μm (Cat#:87003)流 速:0.2mL/min进样量:5 μL 柱 温:35 ℃流动相:A: 10 mmol/L乙酸铵 B:乙腈梯度设置 时间/Min. 0 4 4.5 6 6.5 12 A(%) 80 50 10 10 80 80 B(%) 20 50 90 90 20 204.2 质谱条件:电离模式:ESI 扫描方式: 负离子扫描检测方式:多反应监测 电喷雾电压:-4500 V雾化气压力:50 psi 辅助气压力:50 psi气帘气压力:20 psi 离子源温度:500 ℃定性离子对、定量离子对、碰撞气能量及去簇电压见下表http://ng1.17img.cn/bbsfiles/images/2015/07/201507211549_556504_1610895_3.jpg5、实验结果 草鱼、猪肉、牛奶、蜂蜜样品中氯霉素类药物的LC-MS/MS检测添加回收结果 http://ng1.17img.cn/bbsfiles/images/2015/07/201507211552_556505_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507211605_556507_1610895_3.jpg氯霉素多反应监测色谱图http://ng1.17img.cn/bbsfiles/images/2015/07/201507211605_556508_1610895_3.jpg甲砜霉素多反应监测色谱图http://ng1.17img.cn/bbsfiles/images/2015/07/201507211605_556509_1610895_3.jpg草鱼空白样品总反应监测图http://ng1.17img.cn/bbsfiles/images/2015/07/201507211606_556510_1610895_3.jpg草鱼加标样品总反应监测图http://ng1.17img.cn/bbsfiles/images/2015/07/201507211606_556511_1610895_3.jpg猪肉空白样品总反应监测图http://ng1.17img.cn/bbsfiles/images/2015/07/201507211606_556512_1610895_3.jpg猪肉加标样品总反应监测图http://ng1.17img.cn/bbsfiles/images/2015/07/201507211606_556513_1610895_3.jpg牛奶空白样品总反应监测图http://ng1.17img.cn/bbsfiles/images/2015/07/201507211607_556514_1610895_3.jpg牛奶加标样品总反应监测图http://ng1.17img.cn/bbsfiles/images/2015/07/201507211607_556515_1610895_3.jpg蜂蜜空白样品总反应监测图http://ng1.17img.cn/bbsfiles/images/2015/07/201507211607_556516_1610895_3.jpg牛奶加标样品总反应监测图
请教安捷伦[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url] 6470检测动物源性食品中氯霉素质谱条件的设置
各位大神: 刚开始接触动物源性食品中硝基呋喃类代谢物检测工作,求优化好的前处理步骤[img]https://simg.instrument.com.cn/bbs/images/brow/em61.gif[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP