测四氟丙醇中的杂质叔丁醇的含量。因为现有两台[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]GC979/GC1690,手动进样0.8UL,都是FID。柱子都是兰化所的1701毛细柱。分析同一个样品,总峰面积1690为1080W,9790的为998W,结果1690那台叔丁醇的含量是9790的两倍,做了3次试验都是一样情况。两台色谱的载气,空气,氢气的流速都一样。不知道怎样才能确定哪个是准确的。是不是用内标?本人接触色谱不久对内标不是很懂,对叔丁醇选什么内标物好?内标物是不是要特别买?我看试剂的含量都是个范围。
最近要测定原辅料磷酸三丁酯(TNBP)的含量,可我不知道怎么测定,看到药典上用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]做的,但我又从来没做过[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],所以也不清楚,而且药典上的步骤是测样品中的TNBP残余量的,这个原辅料的TNBP含量又很高(〉98%),我不知道该怎么测定,请哪位高人指点一下,给个好方法把,谢谢了。
我实验室要按国标分析化学试剂磷酸三丁酯(TBP)的含量,国标方法是气相色谱,但是含量测定国标上写的不是很清晰,只给了检测条件,定量是归一法,没有详细的操作步骤(比如是否要对化学试剂磷酸三丁酯(TBP)进行前处理,是否可以直接进样等),要是有做过这方面的或是对气相经验丰富的,介绍一下经验啊,你们是怎么做的啊,主要是操作的步骤,谢谢分享啊,附件是国标。
腐植酸铵附录法测腐植酸含量要加上无机盐加以校正求其腐植酸,无机盐会有哪些呢
有机化工产品精馏过程副产有丁醇水溶液,取样发现取样瓶内很快就分层了,除了水,丁醇外,还有少量甲醇杂质。[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]有啥办法准确定量其中的丁醇含量。
我做的是一种碳酸盐岩中氟元素含量的测定,实验室的氟电极极不稳定,清洗多次也达不到所需要的370mV的标准,总是持续的下降,请教一下如何才能让氟电极的读数趋于稳定?或者还有别的方法能来完成这个测定?急需,先谢谢各个大哥大姐了
利用激光拉曼光谱法是否可以快速检测油品(航煤、汽油、柴油)中的水份和杂质含量?
最近我从客户那里接了一批样品,其脂肪含量很高,要求按国标5009.18第三法测定其中氟含量。因为第一次做这类样品,没有经验,请大家多指点。1)按国标所描述的氟电极不适合脂肪高样品,那么有什么好办法对其进行样品预处理?灰化(会不会有氟损失)?2)客户说氟含量可能很低,小于1ug/g,这样的浓度下用标准加入法还是标准曲线法比较准确?3)国标第三法中标曲溶液浓度范围 0.02~0.2 ug/ml,大家有试过在此条件下做过吗?所得的标曲斜率应该是多少?按照电极性质,浓度没差10倍,斜率应该在58加减2mV?谢谢大家!
有色行业标准中关于氟化盐中氟含量的测定是用蒸馏—硝酸钍容量法,可是在实际操作过程中,这套蒸馏装置的配套制作和操作都比较繁琐,我们目前使用氟氯化铅的方法,但是氟化铝中的氟含量测定有时超过允许差,不知道有没有测定氟化铝中氟含量的同仁,有没有更好的办法?
各位大侠帮帮忙!!最近在做一种含氟树脂,需要测主体和表面的氟含量(不需要表示总的氟含量),常用的XPS好像测试的深度只有10nm,EDS好像可以达到1微米,不过不知道哪个的准确度高,还有没有其他的一起来表征氟元素的含量(表面和主体)!!
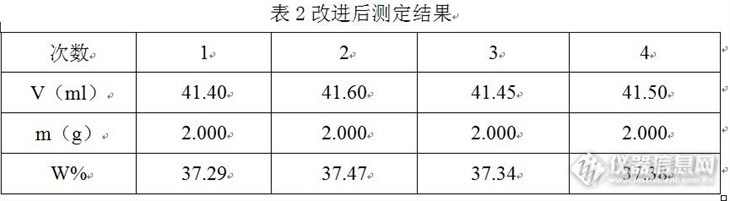
氟硅酸含量的测定失败的探讨和改进摘要:本文对采用HG/T 2832—2008进行氟硅酸测定过程遇到的问题进行简单的说明,就过程中出现问题的现象进行简单的阐述,对问题产生的原因进行了分析,最后结合本人的经验对现有方法进行了局部的改进,获得良好的结果,获得较好的精密度,有利于大家以后在做类似样品的时候能够借鉴和学习。希望大家在工作中能做到举一反三思路,解决一些常见的问题。关键词:氟硅酸、测定、改进氟硅酸易分解为四氟化硅和氟化氢。水溶液无色,呈强酸性反应。有腐蚀性,能侵蚀玻璃。氟硅酸有消毒性能,用于制氟硅酸盐和冰晶石,并用于电镀、啤酒消毒、木材防腐等。应朋友要求帮忙测定其样品中氟硅酸其含量,按照标准方法且结合咱实验室的条件进行测定,但效果不好,精密度较差。怎么解决呢???最后结合个人经验对方法的理解,对方法进行改进,精密度非常的好,说明方法的改进实验非常的成功。现与大家分享一下。1 国标方法简要氟硅酸与硝酸钾反应,生成氟硅酸钾沉淀和硝酸,先在低温下以氢氧化钠标准滴定溶液滴定反应生成的硝酸及其他的酸(微量的HF)。然后滴定经沸腾水解产生的氢氟酸。根据滴定后者时氢氧化钠标准滴定溶液的用量计算出氟硅酸的含量。主要的反应方程式如下:H2SiF6+2KNO3= K2SiF6↓+2HNO3HNO3+ NaOH=Na NO3+ H2OHF+ NaOH=NaF+ H2OK2SiF6+ 4NaOH=2KF+ 4NaF+SiO2↓+ 2H2O2 本次实验涉及的试剂硝酸钾饱和溶液。氢氧化钠标准滴定溶液:C(NaOH)=0.5mol/L酚酞指示液:10 g/L。溴酚兰指示液:2%(乙醇),KCl-乙醇洗液:1+13 分析步骤称取2.000g试样,精确至0.0002g,置于250 mL锥形瓶中,加入10 mL饱和硝酸钾溶液和10 mL水,于冰箱室中放置30 min。取出加人3滴酚酞指示液,用氢氧化钠标准滴定溶液滴定至红色保持30 S不褪。将溶液加热至沸,将滴定管调零后用氢氧化钠标准溶液滴定至稳定的红色为终点,记下消耗的体积(V)。4 结果计算氟硅酸含量以氟硅酸的质量分数计,按公式计算:http://ng1.17img.cn/bbsfiles/images/2013/07/201307052306_449753_1607403_3.jpg式中:[size=12pt
[em0912]最近在修改化验的操作规程,发现一种原料的含量是用氢氧化钠滴定的,写的很简单,只说是平行两份。按照2005年版的药品检验操作规程,对紫外测定含量,液相测定含量等等都有明确的数量和RSD要求,但是关于滴定的我没有找到,求助各位,有没有什么标准是用于含量滴定的 一般最少要求几份,RSD 要求多少呢 ,谢谢了[em0909]
请问在做防腐剂含量测定时,系统适应性试验该怎么做?或者需要做系统适应性吗?因为这个测防腐剂的色谱条件与我测主药含量测定的条件不一样。如果我在测主药含量时做了系统用适应性(含有主药和各种杂质),那么换了条件之后的系统适应性只单做含有两个防腐剂的就行了吗?
测药品含量时对其中一个辅料氯化钠含量测定时,用的硝酸银滴定,按照中国药典的方法,算出来的结果为120%,考虑可能空白影响较大,滴定空白发现空白的终点难以控制,溶液透明,滴定终点消耗体积较大,扣除空白所得含量更大。有没有其他方法测含量的,谢谢!
拉曼做金属氧化物含量的下限是多少? 我有一几种氧化物的混合物,其中MoO3含量只有5%,XRD检测不到,拉曼可以吗?初次接触拉曼,请见谅!
氩气中氮气含量的判定一般市售的氩气中,或多或少含有氮气(液氩会好些),而氩气中氮气含量的多少,直接影响着“引燃”的难易,因此,对氩气的验收是一项非常重要的工 作。使用纯度合格的氩气“引燃”等离子体后,将输出氩气切换至待检查气瓶,高纯去离子水进样,运行全谱扫描(短波)。如图7所示,氮气光带越亮, 说明氮气含量越高(高纯氩气仅仅出现4个氮谱线的亮斑,为N174.272nm{493}、N174.272nm{494}、N 174.525nm{493}、N174.525nm{494},不会出现亮的谱带)。气绝缘性能变差而影响仪器正常工作。另外,位于等离子体屏蔽室左上方的光室石英端窗长时间暴露在外界气氛中,也难免会沾附污物,影响仪器的分析灵敏度。因此,清洁工作是仪器维护保养中一项不可或缺的重要工作。
有谁知道对羟基苯甲酸丁酯钠和对羟基苯甲酸丙酯钠的含量测定方法,急!

纺织品成分分析中含量1%以下与微量纤维怎么界定与判定[font='times new roman'] 纺织品纤维成分分析是[/font][font='times new roman']根据纺织纤维[/font][font='times new roman']的外观[/font][font='times new roman']纵[/font][font='times new roman']截面和横截面的形态特征和内在的不同性质,采用物理方法或者化学方法,辨别和区分各种纤维。通过各种实验来鉴别各种纺织纤维[/font][font='times new roman']。[/font][font='times new roman'],不仅用于单一纤维的定性,还用于鉴别及定量多种纤维混纺的纺织品[/font][font='times new roman']的纤维组成。[/font][font='times new roman'] 在常见的纺织纤维中,大多数都有了比较成熟的定量方法,比如[/font][font='times new roman']GB/T2910-2009[/font][font='times new roman']系列的检测方法,常规的纤维定量基本都能用到,其纤维定量的方法也比较成熟了,基本上按照纤维定性的结果选择适合的[/font][font='times new roman']检测方法进行检测即可。[/font][font='times new roman'] 纤维成分分析一般是取两个平行样,两个测试[/font][font='times new roman']样一起[/font][font='times new roman']进行前处理,需要褪色处理的要进行褪色处理,然后进行恒重,选择合适的分析方法溶解,干燥平衡,最后进行计算,两个平衡样的结果偏差不超过[/font][font='times new roman']1%[/font][font='times new roman'],即求两个试样的平均值为测试的最终结果。上报结果,成分分析完成。[/font] 但是最近遇见几个纤维计算后其中一种纤维含量再0.7-0.9%之间,均小于1%,这个值是按照标准溶解方法化学定量出来的,按照标准方法GBT 29862-2013纺织品 纤维含量的标识,进行出报告的话那么我这个样品成分定量结果是:50%聚酯纤维,49.3%棉,0.7%粘纤,按照标准方法检测和标示,我这个都没有任何问题,肯定也不算错,也是没有问题。 当时考虑到人员误差,试剂误差等等原因,最终把同一块样品送到省纤维检测院和市级纺织服装检测中心,省纤维检测院出的报告为50.5%聚酯纤维,49.5%棉(含微量其他纤维);市级纺织服装检测中心出的结果为:50%聚酯纤维,50%棉. 为了搞清楚他们的测试原理和方法是否和我们一至,经过多方努力终于联系到具体做这个适试样的两个工程师,省纤维检测院的工程师经确认我就是这个样品的送样人时,告诉我,如果按照溶剂法,几乎就没有微量纤维,两个试样溶解误差都不止0.5%,所以溶解微量纤维超过0.5%很正常的,哪怕这个纤维没有粘纤,你按标准进行溶解也会有百分之零点几的数据出来,所以一个试样溶解不超过1%的数据结果都是不可信的结果,一般都是出微量纤维,这个不是标准,是经验。 市级纺织服装检测中心的工程师告诉我,他们在显微镜下一个工程师能看到有1根粘纤,另一个工程师在显微镜下没有看到有粘纤,这个一般直接可以判定是微量,而且是不均匀的,按照[img=,636,103]https://ng1.17img.cn/bbsfiles/images/2021/08/202108031137589618_4128_2154459_3.png!w636x103.jpg[/img][font='times new roman'] 直接[/font][font='times new roman']出的结果为:[/font][font='times new roman']50%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']50%[/font][font='times new roman']棉[/font][font='times new roman'].[/font][font='times new roman'],这个是没有任何问题的。[/font][font='times new roman'] 通过这个样品,我们实验室内部也专门制定了一个作业程序,并进行了一个培训[/font][font='times new roman'],对成分分析做了以下几点[/font][font='times new roman']分析[/font][font='times new roman']。[/font][font='times new roman']1. [/font][font='times new roman']这个样品成分定量结果是:[/font][font='times new roman']50%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']49.3%[/font][font='times new roman']棉,[/font][font='times new roman']0.7%[/font][font='times new roman']粘[/font][font='times new roman']纤[/font][font='times new roman'];[/font][font='times new roman']50.5%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']49.5%[/font][font='times new roman']棉(含微量其他纤维)[/font][font='times new roman'];[/font][font='times new roman']50%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']50%[/font][font='times new roman']棉[/font][font='times new roman']三个结果都是正确的,[/font][font='times new roman']都不算错,但是结合实际情况,认为这个[/font][font='times new roman']50.5%[/font][font='times new roman']聚酯纤维,[/font][font='times new roman']49.5%[/font][font='times new roman']棉(含微量其他纤维)[/font][font='times new roman']是最合理的结果。[/font][font='times new roman']2. [/font][font='times new roman']成分分析定性要区多个试样,因为可能存在不均匀性,特别是纤维含量比较少的情况下[/font]3. GBT 29862-2013纺织品 纤维含量的标识,要多理解其中的说明,只要是符合其中的要求,就是可以的。
我用碘氟法测铜含量时,滴定到终点溶液变成乳白色以后总是反色,就是呆上一分钟以后就又回变到蓝紫色了,不知这是为什么,请指教,非常感谢。
请教丁二酮肟法测定镍含量时,怎么控制pH值呢,用精密试纸还是pH计啊,做过镍含量检测的帮帮忙
豆制品中蛋白质含量丰富,发酵类的食物更好,比如豆豉、腐乳、发酵的臭豆腐,纳豆,发酵类的食物富含维生素B12。
我合成了含巯基的纳米材料,想用ellman试剂测一下含巯基的量。采用定点测量,建立紫外标曲,然后测样品,得到样品的吸光度,代入就得到了样品的巯基浓度吗?还有一个问题,我用另一个已知浓度样品测了一下巯基含量,结果显示比我配的巯基浓度低了近一半,是我放置反应时间较短吗?
按照GBT 22104-2008用离子选择性电极测定土壤中氟离子含量,移取的标液体积分别是0.00 0.50 1.00 2.00 5.00 10.0 20.0 ml,然后以毫伏数(mV)与氟含量(ug)绘制对数标准曲线,有一个问题,lg0没有任何意义,这个数字根本就是不存在的,怎么弄?之前只做过水质中氟离子含量、大气中氟离子含量,在做曲线的时候都没遇到这个问题,在土壤中却有了。

零零散散做了几次原子吸收,检测土壤中的重金属含量,问一个比较low的问题。如果试样为标准物质,其中Pb含量为25mg/Kg(即0.0125mg/0.5g),取样0.5g,消解时加入5mL氢氟酸。贴了两张图,第一张AR级别的氢氟酸,第二张GR级别的氢氟酸。我理解的图片中重金属杂质的含量(以Pb计)为Xg/100g,那么5mL氢氟酸中含有的重金属(以Pb计)分别是AR:0.025mg,GR:0.005mg,分别是试样中Pb含量的200%,40%。不知道这样计算是否有误(杂质全部以Pb计算),如果无误,试剂重金属杂质对测定结果有一定的影响。http://ng1.17img.cn/bbsfiles/images/2015/11/201511131312_573404_2913979_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/11/201511131312_573405_2913979_3.png
如题:今天做污水硬度(滴定法),发现其中结果钙的含量比镁大,而且大很多。师傅说做有问题,说结果一定是钙含量大于镁含量。后来又重新做了一遍,结果相差不大,不知道师傅这种说法对吗?同一水质中钙的含量一定比镁大吗?
HPLC法测定郁福来胶囊中芦丁的含量,Inertsil ODS-3.5μm 150×4.6mm色谱柱, SecurityGuard Guard Cartridge Kit保护柱,乙腈-1 %甲酸(19∶81)流动相,检测波长360 nm,柱温30℃, 线性范围为0.219~2.19μg.mL-1,r=0.998,平均回收率为99.2%,RSD为1.52%。
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=31408]高含量银杏萜内酯富集方法研究[/url]
2,4-D二甲胺盐中存在的是2,4-D酸吧,我们可以2,4滴丁酯标样分析它吗,此样品中有2,4滴丁酯没有?如果没有2,4滴丁酯可以分析2,4D二甲胺盐中2,4d酸的含量吗
请问各位老师: 乙酸乙酯中的叔丁胺测定含量的方法,简单易操作的。谢谢
按照药典检测氟含量,加入指示剂后没有出现蓝紫色,而是黄色,实验现象错误,是什么原因引起的呢?







 我要推广仪器
我要推广仪器
 下载APP
下载APP