荧光分光光度计更换灯后,测量值由130多降到40多,什么原因呢?测定条件都没变化
岛津240AAS测镉5mg/l的吸光度达到0.9多,什么原因呢?从准确度的角度考虑,吸光度不是要在0.2~0.8之间吗?求路过的大神朋友们指点一下。
最近做一个化合物,分子量1100多,走MS想确定是不是我要的东西,发现一到我的主峰位置MS就出倒峰,请问是什么原因,如何解决?谢谢!
气相的色谱图基线毛刺多,大概的原因是什么呢?如何修正
[color=#0021b0][size=5]各位,我想请问,我这次出了一个倒峰,以前的没有的,不过我通过手动积分使我要的那个峰正常积分算出结果了,可是我想知道倒峰出现的原因是什么?如何解决?对结果有影响吗?[/size][/color]
询问---GPC的 尾峰比较高 达到200多 ,什么原因?询问---GPC的 尾峰比较高 达到200多 ,什么原因?询问---GPC的 尾峰比较高 达到200多 ,什么原因?
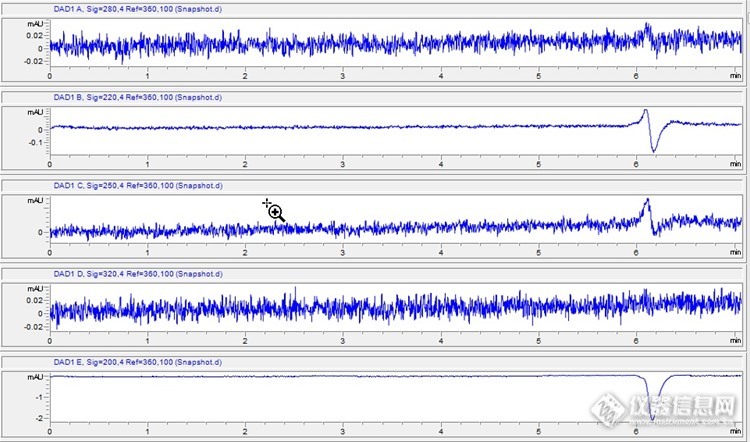
等度洗脱,流动相0.004M硫酸水溶液。样品纯水,进样量10微升。在6分钟时出现一个倒峰,不同的检测波长,信号形状不同,请问出现这种信号的原因有哪些?另外RI检测器上是一个倒峰,形状和200nm的类似个人感觉:RI倒峰是因为水的通过,和参比相比,水的折光更小,所以出现负值DAD相似时间出现负值,是不是也是因为样品水的原因呢?所以这个位置并不是污染物?http://ng1.17img.cn/bbsfiles/images/2016/06/201606031407_595911_1654762_3.jpg非常感谢
大肠菌群、大肠杆菌检测时,所用培养基LST、BGLB、EC肉汤等这些需要用小倒管(杜氏管)考察产气的培养基,灭菌后,偶尔会有小气泡留在小倒管内,导致培养基不能使用。重复灭菌不仅费时,更重要的是会破坏培养基的有效成分。为了避免这类问题的发生,我公司实验室根据实验经验,做出以下几种可能原因分析:原因一:高温高压灭菌锅工作压力不够,使气体不能完全排出高压锅在灭菌过程中产生的最大压力不够,不能将小倒管内的气体挤出,导致灭菌后任留有小气泡。此外,高温高压灭菌锅若是温度上升缓慢或者达不到灭菌温度,也会出现类似现象。解决办法:使用检查灭菌效果的用品-压力蒸汽灭菌指示卡,对高温高压灭菌锅的灭菌情况进行监测。原因二:试管塞塞得太紧在灭菌时,试管塞塞得太紧(有些硅胶塞与试管不配套)会导致试管内压力与灭菌锅内压力不一致。当灭菌锅升温升压时,锅内压力会大于试管内压力,试管内压力不够,不能将小倒管内气体排尽,就留有气泡;当灭菌锅降温降压时,锅内压力小于管内压力,可能会使试管塞和培养基崩出,培养基报废。解决办法:改用塑料帽、不锈钢管帽,或者与试管相匹配的硅胶塞。原因三:小倒管口太小小倒管在加热加压后,一方面液体培养基要进入小倒管,另一方面小倒管内部的空气要被排出,如果小倒管管口太小,液体不容易进去,里面的空气不容易被排出,就会导致有小气泡生成。解决办法:改用开口比较大的倒管。 以上提供几种可能形成气泡的主要原因,还有几点建议:1.可以在高压锅压力降下来后,温度不太高的时候将培养基从锅中拿出,迅速放在冷水中,急冷,注意要使冷水高度超过试管中液面高度。利用热胀冷缩的原理,可以使小倒管中气泡排出。2.若使用非全自动高温高压灭菌锅(此类锅一般需要在100℃手动关闭放气阀),则不要在锅刚达到100℃时就关闭放气阀,可以让高压锅在100℃时多排一会儿热气,这样有助于小倒管中气体排出。此外,尽量不要采用将灭菌后的试管颠倒或大角度倾斜的方式,使小倒管内气体排出。此种方法可能会导致培养基通过试管口或试管塞受到污染,或者导致试管内液体流出。
测羊奶中砷.汞含量是标准空白与样品空白都是4000多我用的AFS-2202E双道原子 荧光光度计 刚刚学习使用不久的新手,最近在测羊奶中砷.汞含量是标准空白与样品空白都是4000多,洗了一天液洗不下去是什么原因呢?我也用它测铅,测砷汞的载液是5%硝酸 ,测铅用的是2%盐酸,盐酸含汞是否是它影响的呢?有什么办法避免吗?
手动进样,一直是倒峰,虽然是倒峰的重现性非常好,可能的原因是什么(仪器比较老)
有用户反应盐雾腐蚀试验箱有时喷雾正常,可是却出现了盐水自动增多的现象,不知道这是为什么?下面雅士林仪器就这方面问题做出简单直观的阐述,希望能为用户解答疑问! 找出盐水自动增多现象出现的可能原因,有两种可能性,其一,向水槽加水时加到了盐水槽中;其二,盐水槽与水槽之间出现了漏洞,水浴的水漏到了盐水槽。放干盐水,将水槽装上水,看是否有漏水问题。解决办法,先将盐水槽彻底擦干检查水的来源。 以上内容就是关于盐雾腐蚀试验箱关于盐水突然自动增多现象的简单阐述,如需了解更多内容,欢迎访问雅士林仪器官网:盐雾腐蚀试验箱盐水自动增多的原因?
用岛津GC-2010测定CH4时发现,样品测定时出现大的倒峰,峰高可以达到4000 uv 左右,有时甚至比样品的正峰大,可是同一条件下进入标准样其倒峰明显变小或几乎没有。请大侠们赐教其原因
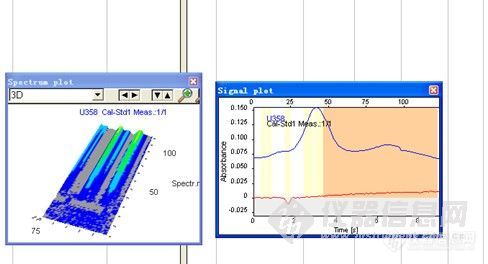
各位专家,在摸索U的分析条件时,好不容易出现了一个峰,却是倒峰。而且背景比信号高,这是什么原因呢?可以从哪些方面进行优化啊?希望大家能够指导一下,谢啦!http://ng1.17img.cn/bbsfiles/images/2012/06/201206261656_374534_2374311_3.jpg
北京吉天AFS-8220原子荧光测汞试剂空白1000多,样品都是-300多,什么原因?一直用的同一批酸,管路换的新的
我是一枚初次用液相的小白,我用的流动相:0.5%乙酸?100%乙腈按照9:1的比例溶解标品,做出来的曲线出现倒峰,也不知道是什么原因造成的?求各位大神指导![img]https://ng1.17img.cn/bbsfiles/images/2019/04/201904181942013874_6018_3893587_3.jpeg[/img]

http://ng1.17img.cn/bbsfiles/images/2014/05/201405271122_500474_2774518_3.jpg各位,最近做zek18种多环芳烃时发现,中间PY出两个峰,前边萘和最后的几个组分都是一个峰,以前也没有这种情况。后来我以为是进样量太大了,从1微升减到0.5,PY变回一个峰了,但现在又出现这种情况,可能是什么原因。进样方式是脉冲不分流
最近进行试验建立一个分析方法,流动相是甲醇-水,使用梯度洗脱,但是在分析物峰的前面紧邻着出现一个倒峰,不知道是什么原因,请各位分析一下,都有哪些原因?
请教各位前辈,出现倒峰是什么原因呀?检查了柱子、流动相都没有问题,这是怎么回事儿呢?
大家在做液谱时,可能都碰到过各种各样的鬼峰吧!我最近就经常遇到倒峰。导致倒峰的形成有哪些原因呢?欢迎大家来交流哈~~~
各位液相高手好!我做的是毛细管区带电泳拆分手性药物,为什么出现了一正一倒峰,而且这两个峰是紧挨着?谢谢说明原因。
待基线走平衡后,进样品,溶剂峰后出现一个小倒峰,是什么原因?走好几针,都出现同样的状况。是不是系统里面有气泡的原因?流动相有经过超声,管道上也没看到有气泡。
各位液相高手好!我做的是毛细管区带电泳拆分手性药物,为什么出现了一正一倒峰,而且这两个峰是紧挨着?谢谢说明原因。
ECD是否适合多溴联苯醚的检测?原因?大侠们,尽量深刻点!!!
ECD检测器出倒峰是什么原因?溶剂峰出现倒峰先是出的正峰到了尾点突然出现倒峰现象,而样品峰确实完全正常的。这是什么原因造成的呀!
最近多地出现大雾天气,主要原因是什么?天瑞有气体检测方面的仪器吗?
昨天他们反映谱图全部为倒峰,但是积分面积没错,不知是什么原因,
我做的药品在PH=7的时候目的时间出现来倒峰,但是在PH7都不会出现倒峰,可以得到目的峰,请问各位是什么原因呢?
我用万通792型[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]做样时出现了好几个倒峰,这是什么原因?还请各位专家指点迷津啊!
做的是多环芳烃EPA16,目前有[font=&]氘代内标(菲-D10,芘-D12),想用菲-D10(3环)来校正2-3环的多环芳烃(在开始时加入,每次计算回收率校正数据),用芘-D12(4环)来校正4-6环多环芳烃,请问这种做法可以吗?各种氘代多环芳烃内标具体可以监测那些多环芳烃呢?有无明确要求呢?是每一个环数的多环芳烃都要有氘代内标吗?(高环数氘代内标太贵了)[/font]
开仪器时,发现抑制器的背景电导从20,几秒钟内涨到2000多,吓坏了。赶紧将抑制器关掉,检查没有漏气现象(曾经有过漏气,背景电导也很高),使用的是ICS-1500戴安公司的,RFC-30外加水抑制器,会是什么原因导致???







 我要推广仪器
我要推广仪器
 下载APP
下载APP