赛默飞:蛋白质组学技术在病毒感染致病机制的亮点研究
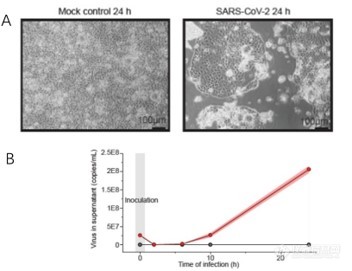
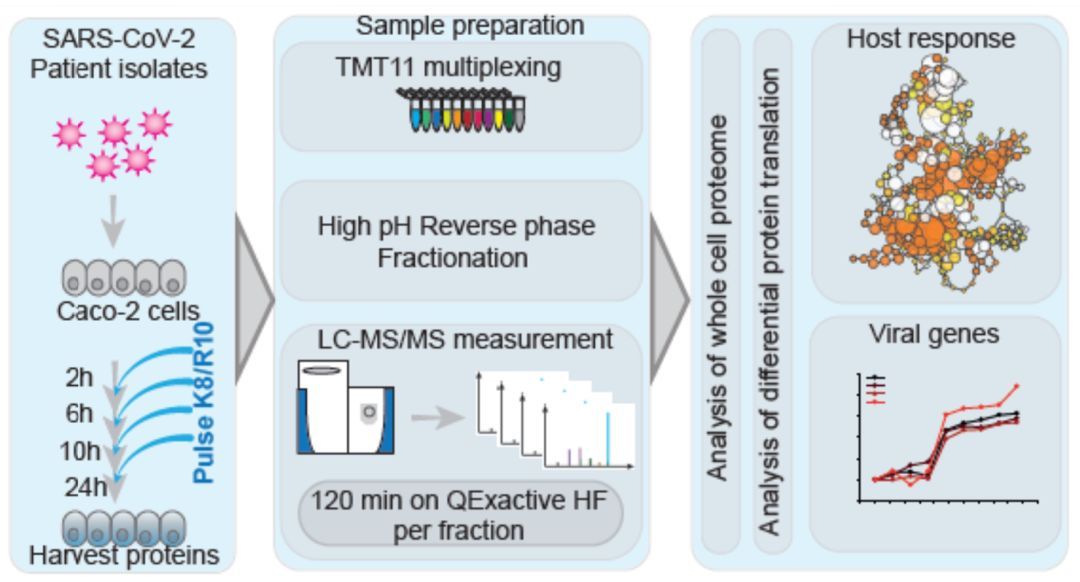
p style=" text-align: justify line-height: 1.75em text-indent: 2em " strong 仪器信息网讯 /strong 2020年,新型冠状病毒肺炎在中国和国际的迅速传播引发了全球卫生紧急情况。仪器信息网在密切关注疫情发展态势的同时,也更加关注病毒感染的致病机理等相关研究进展。近年来,组学研究成为生命科学基础研究领域的重点,对于病理、毒理学、药物动力学等具有重要价值,相关高水平学术期刊大量报道了科研人员利用组学技术开展的病毒致病病理学的研究成果,也对于此次疫情的进一步研究具有一定参考意义。 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 基于此,仪器信息网推出了 a href=" https://www.instrument.com.cn/zt/omics2020" target=" _blank" span style=" color: rgb(0, 112, 192) " strong “组学技术在病毒感染致病机制中的亮点研究及技术进展” /strong /span /a 专题,为广大业内专家及用户介绍基于蛋白组学或代谢组学等多组学技术在病毒感染致病机制中的研究应用及技术进展,增强业界专家与仪器企业之间的信息交流,提供更丰富、更专业的技术文章,谨以此致敬所有奋战在抗疫一线的白衣天使以及幕后深耕的研究学者。 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 从2019年底由SARS-CoV-2引起的新型冠状病毒肺炎(COVID-19)具有高传染性,新型冠状病毒肺炎在中国武汉的出现及其在中国和国际的迅速传播引发了全球卫生紧急情况。住院患者的总死亡率在2.3%至11%之间。本文介绍了 strong 基于蛋白组学或代谢组学等多组学技术在病毒感染研究的应用及技术进展 /strong 。 /p p style=" text-align: center line-height: 1.75em text-indent: 2em " span style=" color: rgb(192, 0, 0) " strong 1.靶向蛋白定量方法研究新冠病毒感染蛋白动态变化 /strong /span /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " strong 1.1 Orbitrap Eclipse高灵敏度平行反应监测靶向定量新冠病毒蛋白 /strong /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 最近,研究人员通过使用了赛默飞Orbitrap Eclipse检测SARS-CoV-2病毒蛋白肽段序列,作为潜在的诊断工具,利用easy1200液相和Eclipse质谱使用靶向方法-平行反应监测(PRM)追踪病毒感染Vero细胞样本中几种SARS-CoV-2蛋白的肽段变化。 span style=" text-indent: 2em " 作者最初研究了SARS-CoV-2蛋白PRM实验的检测限,主要是核衣壳(NCAP),这是最丰富的病毒蛋白,因此是比较好的候选测试项目。 /span span style=" text-indent: 2em " Orbitrap Eclipse 的PRM结果显示对该蛋白灵敏度可达到amol级别(大约为0.9pg)。粗略计算表明,这种灵敏度水平应足以检测理论上与约10000个SARS-CoV-2颗粒相对应的蛋白质量。其他SARS-CoV-2蛋白也被发现是PRM靶向方法的良好候选蛋白。 /span /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " span style=" text-indent: 2em " (参考文献:https://www.biorxiv.org/content/10.1101/2020.04.23.057810v1) /span /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 417px " src=" https://img1.17img.cn/17img/images/202004/uepic/3b037340-0003-46f0-a617-942d85139408.jpg" title=" 图片 1.png" alt=" 图片 1.png" width=" 600" height=" 417" border=" 0" vspace=" 0" / /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp & nbsp 图. Orbitrap Eclipse靶向PRM监测SARS-CoV-2病毒蛋白肽段序列 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 此外,作者还进行了整体蛋白质组学分析,在60-90分钟分析时间内,共鉴定出6500种相关蛋白质。这表明蛋白质组学可以用来研究病毒感染宿主细胞的蛋白质组,也是未来新兴的研究工具。 /p p style=" text-align: center line-height: 1.75em text-indent: 2em " span style=" color: rgb(192, 0, 0) " strong 2.蛋白质组学研究新冠病毒感染的调控机制研究 /strong /span /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " span style=" color: rgb(0, 0, 0) " strong span style=" text-indent: 2em " 2.1 Orbitrap 高分辨质谱研究病毒宿主相互作用蛋白揭示感染机制 /span /strong /span /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 另外一篇研究关于SARS-CoV-2病毒的基因组有29811个核苷酸,编码28-29个蛋白质。它编码的蛋白质之一是蛋白质E,它是一种包膜蛋白,一种有助于形成病毒油泡的结构蛋白。一旦病毒进入细胞,它还将有其他的功能,例如,它附着在蛋白质上,有助于打开和关闭其基因,当E蛋白干扰时,模式可能会改变。 span style=" text-indent: 2em " 据报道,蛋白质E与寄主细胞中的溴多胺蛋白结合,Bromodomain 4(BRD4)是BET亚家族蛋白,通过识别乙酰化组蛋白提供了一个招募平台作用,与非组蛋白靶点相关,除与病毒蛋白复合外,还参与NFKB信号转导、Myc调控。此外,BRD4还与其它bromodomain蛋白存在协同作用,其中一些还可能执行泛素化功能。 /span /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 另有一位作者使用QE HFX质谱描述了BRD4相互作用蛋白的分子表型,使用了经BET抑制剂、与BRD4结合的化合物以及可能的其他bromodomain蛋白处理的人类B细胞系蛋白质组。已鉴定的蛋白定位于SARS-CoV-2病毒感染细胞的相互作用重编程途径和复合物中,如下图所示。(A proteomic model of SARS-COV2 infection by comparing the interactomes of BRD4 with BET-inhibition and SARS-COV2 viral proteins – implications for re-purposing approved drugs or ubiquitin-mediated degradation of select candidates) /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 422px " src=" https://img1.17img.cn/17img/images/202004/uepic/74611d77-e12d-4700-b696-f209d44d255a.jpg" title=" 图片 2.png" alt=" 图片 2.png" width=" 600" height=" 422" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图.蛋白组学分析SARS-CoV-2病毒感染细胞后BRD4相关互作蛋白 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " & nbsp strong 2.2. Q Exactive HF质谱非标定量技术监控病毒感染细胞实时变化 /strong /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 另一篇研究通过使用Q Exactive HF质谱进行鸟枪法蛋白质组学研究,重点是获得SARS-CoV-2感染的相关信息,确定病毒颗粒抗原产生的最佳条件。 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 为了做到这一点,作者用SARS-CoV-2感染Vero E6细胞(MOI 0.01和0.001)。然后他们用LFQ(非标记定量方法)方法在几天内监测感染的实时变化。他们鉴定了3220个Vero细胞蛋白和6个SARS-CoV-2蛋白,其中细胞27388个和病毒94个特异肽段(FDR& lt 1%)。病毒蛋白水平在不同时间点的动态变化表明,SARS-CoV-2蛋白合成在感染后持续增加,在感染第3天左右达到最大值。为了评估MS获得的病毒图谱在多大程度上反映了病毒的产生,作者通过qPCR在同一时间点测量了SARS-CoV-2 RNA分子。他们观察到最丰富的病毒蛋白产量的变化,反映了SARS-CoV-2 RNA分子数量的变化,证实可以使用基于MS的LFQ非标定量监测SARS-CoV-2感染动力学。 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 参考文献:(https://www.biorxiv.org/content/10.1101/2020.04.17.046193v1) /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 352px " src=" https://img1.17img.cn/17img/images/202004/uepic/edfb0017-9692-44d5-877c-18107180805b.jpg" title=" 图片 3.png" alt=" 图片 3.png" width=" 600" height=" 352" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图. Q Exactive HF质谱进行鸟枪法蛋白质组学研究SARS-CoV-2感染的相关蛋白 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " strong 2.3 SILAC和TMT结合多重增强蛋白质动力学方法监控转录组和蛋白质组的变化 /strong /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 来自法兰克福大学医学病毒学研究所的Jindrich Cinatl教授和歌德大学医学院的Christian Mü nch教授团队发表的题为“SARS-CoV-2 infected host cell proteomics& nbsp revealpotential therapy targets” 的最新论文。在本篇工作中,作者建立了感染SARS-CoV-2的Caco-212细胞模型,运用一种新颖的多重增强蛋白质动力学(multiplexed enhanced protein dynamicsme,mePROD)方法进行蛋白质组学分析。能够在高时间分辨率下确定转录组和蛋白质组的变化,加速确证病毒致病性相关的生物途径以及寻找潜在的药物靶标。 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 对感染细胞进行蛋白质组学分析,该策略取决于是否有合适的允许病毒感染的细胞培养模型,以及对蛋白质进行时间感染特征分析的敏感蛋白质组学方法。为了探究潜在的抗病毒药物,作者建立了一个针对SARS-CoV-2高度兼容的细胞模型,在病毒感染24小时后就能迅速见到细胞致病作用(cytopathogeniceffect,CPE)(图1A)。 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " span style=" text-indent: 2em " 研究人员在病毒感染细胞后的2h、6h、10h和24h,分别用定量PCR技术测量上清液中的病毒RNA拷贝数,发现感染后SARS-CoV-2 RNA数量不断增加(图1B)。这表明病毒在细胞中经历了完整的复制周期,成功建立了功能性的SARS-CoV-2细胞培养模型,该模型可以用于研究细胞中SARS-CoV-2生命周期的不同步骤。 /span /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 255px " src=" https://img1.17img.cn/17img/images/202004/uepic/74c24851-b71a-4b76-ad3e-48c2c19512b4.jpg" title=" 图片 4.png" alt=" 图片 4.png" width=" 600" height=" 255" border=" 0" vspace=" 0" / /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 323px " src=" https://img1.17img.cn/17img/images/202004/uepic/642389b8-9ba3-40e5-a6e4-5384db45b276.jpg" title=" 图片 5.png" alt=" 图片 5.png" width=" 600" height=" 323" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图.& nbsp SARS-CoV-2 在细胞内快速复制模型。 /p p style=" text-align: center line-height: 1.75em text-indent: 2em " A, 病毒感染24小时后的细胞形态变化 & nbsp & nbsp /p p style=" text-align: center line-height: 1.75em text-indent: 2em " B, 细胞上清液中病毒RNA拷贝数的增加 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 为了确定SARS-CoV-2感染的时间分布,如下图所示,作者用SARS-CoV-2单次感染Caco-2细胞后培养2-24小时,并通过mePROD蛋白质组学的方法,即联用新蛋白代谢标记(SILAC)和串联质量标签(TMT)两种标记方法,进行蛋白差异分析。 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " span style=" text-indent: 2em " 首先,将SARS CoV-2病毒与Caco-2细胞孵育一定时间,在收集样品前两小时,将培养基更换为重标SILAC培养基以标记新合成蛋白质 带有部分SILAC标记的蛋白酶切成肽段,经TMT11plex试剂标记后通过high-pH反相分级,最后借助Easy nLC 1200-Q Exactive HF高分辨质谱平台进行高通量蛋白质组学分析,用以表征宿主和病毒基因表达变化。 /span /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 320px " src=" https://img1.17img.cn/17img/images/202004/uepic/8ae32504-08dc-4038-a16f-50dfdab799cd.jpg" title=" 图片 6.png" alt=" 图片 6.png" width=" 600" height=" 320" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " & nbsp 图.mePROD蛋白质组学分析流程 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 在三个重复中,我们分别定量到大约4,200种和7,000种蛋白质的相对翻译速率和相对蛋白质水平。主成分分析(PCA)表明,在感染6小时后实验组首次从对照组中分离出来;随着感染时间的延长两组之间的差异越来越大(下图)。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 455px " src=" https://img1.17img.cn/17img/images/202004/uepic/b35166bd-e928-43eb-bf4f-efb68c04e26e.jpg" title=" 图片 7.png" alt=" 图片 7.png" width=" 600" height=" 455" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图.PCA分析结果 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 与SARS-CoV–1病毒类似,许多RNA病毒会降低宿主细胞的蛋白合成;但是实验组与对照组的总体翻译率变化不大,仅在感染10小时后最大降低了23%(下图)。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 300px height: 486px " src=" https://img1.17img.cn/17img/images/202004/uepic/0279f4d2-43c0-4ed1-8af4-000f7abb60d5.jpg" title=" 图片 8.png" alt=" 图片 8.png" width=" 300" height=" 486" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图, 不同时间点的整体翻译速率(N=3) /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 作者将用对所有定量到的病毒蛋白的平均水平作为参照,计算宿主蛋白与其的距离,对最相关的前10%的蛋白进行网络分析。结果显示宿主细胞本身翻译模式的广泛重塑,可能解释了如何避免总体蛋白质合成发生重大变化(如下图所示)。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 300px height: 408px " src=" https://img1.17img.cn/17img/images/202004/uepic/00a705a5-e243-4531-af41-7c8c827accb6.jpg" title=" 图片 9.png" alt=" 图片 9.png" width=" 300" height=" 408" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图.前10%蛋白的通路分析结果 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 在本篇研究中,通过对SARS-CoV-2感染对宿主细胞进行定量蛋白质组分析,确定了由SARS-CoV-19感染调节的宿主细胞通路,并揭示了药物抑制病毒在人体细胞中复制的相关靶通路。感染过程中细胞功能发生了重大调整,SARS-CoV-2重塑了翻译、剪接、碳代谢和核酸代谢等重要细胞代谢途径,并对这些途径相关的小分子抑制剂进行测试。结果揭示了SARS-CoV-2的细胞感染情况,并鉴定出抑制病毒复制的药物,这些结果也将指导开发COVID-19的治疗方案。 /p p style=" text-align: center line-height: 1.75em text-indent: 2em " span style=" color: rgb(192, 0, 0) " strong 3.Orbitrap高分辨质谱针对新冠病毒糖蛋白结构研究进展 /strong /span /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " strong 3.1. Orbitarp Fusion质谱结合冷冻电镜解析SARS-CoV-2 S糖蛋白多糖结构 /strong /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 来自CCRC的Robert Woods实验室。通过大量实验生成了在SARS-CoV-2 S糖蛋白上发现的聚糖的三维结构。整个研究基于报道的S蛋白使用赛默飞冷冻电镜cryo_EM 分析3D结构和相关的糖组学数据(利用Orbitarp Fusion 质谱结合分析)。 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 他们进行了分子动力学模拟,以观察多糖的微观异质性对表位暴露的影响。模糊位置为聚糖结构。M9型(绿色),M5型(深黄色),混合型(橙色),复合型(粉色)。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 300px height: 330px " src=" https://img1.17img.cn/17img/images/202004/uepic/def92f5e-20fe-4269-b50d-36aa139f3bf9.jpg" title=" 图片 10.png" alt=" 图片 10.png" width=" 300" height=" 330" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图. SARS-CoV-2 S糖蛋白聚糖结构和多糖微观异质性 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " strong 3.2. Orbitrap Fusion Lumos质谱绘制新冠病毒SARS-CoV-2 S蛋白糖基化图谱 /strong /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 新型冠状病毒(SARS-CoV-2)中的刺突蛋白(Spike蛋白)是一种高度糖基化的蛋白质,是病毒结合和进入宿主细胞的关键调节因子,也是研发抗体及相关药物的关键靶点。该蛋白的位点特异性N-糖基化修饰(包含糖基化位点以及糖链信息)分析对于了解其功能和药物研发具有重要意义。在中国2020年3月,四川大学华西医院科研团队在生物学预印本bioRxiv在线发表文章“Site-specific N-glycosylation Characterization of Recombinant SARS-CoV-2 Spike Proteins using High-Resolution Mass Spectrometry”针对该蛋白的位点特异性N-糖基化修饰进行深入分析,具体实验流程如下图 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 283px " src=" https://img1.17img.cn/17img/images/202004/uepic/0bffa430-0704-4b8e-aa77-7392ed0df0aa.jpg" title=" 图片 11.png" alt=" 图片 11.png" width=" 600" height=" 283" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图.利用orbitrap质谱分析新冠病毒S蛋白N-糖基化流程 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 通过使用Orbitrap Fusion Lumos质谱技术的整合糖蛋白质组学新方法,绘制了新冠病毒SARS-CoV-2重组表达蛋白的所有糖基化修饰位点以及位点特异性的N-糖链组成图谱,如下图所示。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 816px " src=" https://img1.17img.cn/17img/images/202004/uepic/2d19678f-d802-4f1a-9b6e-0b0d883f4334.jpg" title=" 图片 12.png" alt=" 图片 12.png" width=" 600" height=" 816" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图. S糖蛋白位点特异性N糖基化位点修饰谱 /p p style=" text-align: center line-height: 1.75em text-indent: 2em " span style=" color: rgb(192, 0, 0) " strong 4.蛋白质组以及代谢多组学研究新冠病毒感染病人血清生物标志物 /strong /span /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " strong 4.1多组学方法研究新冠肺炎轻重症患者血清中蛋白和代谢物生物标志物 /strong /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 西湖大学生命科学学院与合作团队对新冠肺炎患者血液中的蛋白质和代谢物分子进行系统检测,利用多组学方法研究重症患者的血清中存在多种蛋白和代谢物分子变化(Proteomic and Metabolomic Characterization of COVID-19 Patient Sera) span style=" text-indent: 2em " 并找到了一系列生物标志物,有望为预测轻症患者向重症发展提供导向。 a href=" https://www.medrxiv.org/content/10.1101/2020.04.07.20054585v1" target=" _blank" (Proteomic and Metabolomic Characterization of COVID-19 Patient Sera) /a /span /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 实验对99例病毒灭活处理的血清样本进行了安全处理和质谱分析。根据现行临床诊断标准,这些血样被分为对照(健康)组、普通流感组、新冠感染轻症组、新冠感染重症组。运用QE HF质谱对样本的进行蛋白质组和代谢组分析,对血清样本中的蛋白和代谢物的相对浓度进行了广泛全分析,从而揭示了重症患者体内多种独特的分子调控。 /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 具体流程如下图所示 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 211px " src=" https://img1.17img.cn/17img/images/202004/uepic/268f6506-b9d2-4d2f-bdad-5f084133fc34.jpg" title=" 图片 13.png" alt=" 图片 13.png" width=" 600" height=" 211" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图.蛋白质组学结合代谢组学围绕新冠肺炎患者队列进行分析流程 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 与对照(健康)组、普通流感组和轻症组相比,新冠肺炎重症患者的样本中出现了93种特有的蛋白表达和204个特征性改变的代谢分子。其中50种蛋白,与患者体内的巨噬细胞、补体系统、血小板脱颗粒有关。研究团队还发现,在新冠病毒感染的重症患者体内,有100多种氨基酸及100多种脂质均出现显著减少。见下图。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 458px " src=" https://img1.17img.cn/17img/images/202004/uepic/0d26783c-de7b-44cf-912b-dca3f55ae057.jpg" title=" 图片 14.png" alt=" 图片 14.png" width=" 600" height=" 458" border=" 0" vspace=" 0" / /p p style=" text-align: center line-height: 1.75em text-indent: 2em " 图.COVID-19感染后重症患者体内的巨噬细胞、血小板、补体系统的作用通路 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 基于orbitrap质谱技术和机器学习分析方法,短时间内整合蛋白质组、临床、生物、代谢组、计算等多学科数据筛选出重症患者特征性的22个蛋白质和7个代谢物,有望可以辅助现有的诊断分析手段,实现更精准、高效的治疗。 /p p style=" text-align: justify line-height: 1.75em text-indent: 2em " 新型冠状病毒(2019-nCoV)肺炎疫情来势汹汹,牵动着每一个人的心,在这没有硝烟的抗疫战场上,白衣天使和医学研究者就是勇敢的战士。 span style=" text-indent: 2em " 我们整理了基于Orbitrap超高分辨质谱多组学技术在病毒感染致病机制和亮点研究,谨以此文致敬白衣天使和深耕医学研究的学者们,默默付出,勇敢向前,愿望早日完成胜利。 /span /p p br/ /p p style=" text-align: right " 投稿来源:赛默飞色谱与质谱 /p p br/ /p p br/ /p



















 我要推广仪器
我要推广仪器
 下载APP
下载APP