大家讨论一下有哪些方法可以确定一个化合物是不是单体啊?方法越多越好啊?
[b][color=#c001cb]即日起,本版将不定期举行有奖竞答活动~一般为小常识,小问题或者小意外等等~[/color][color=#013add][color=#f10b00]今日题目:如何判断一个样品是不是单体化合物,或者说,如何判断一个样品是不是纯品?[/color]第一位给出最佳答案者,可获得6分奖励~其他参与讨论者,可获得不多于6分的奖励~所有有奖竞答类贴子,可以一直讨论,没有时间限制~[/color][/b][color=#013add][b][color=#ad8e00]注:为体现公平公正,本版版主不参与竞答~[/color][/b][/color]
如题,请教各位老师,哪里能够查到多氯联苯单体IUPAC编号和化合物名称或结构式的对照表。另外HJ 715-2014标准有没有出正版书籍或者正式文本?找了几个供应商都说没有货。网上只找到发布稿,但是内容似乎有一些错误?
有机氟化合物organic fluorine compound有机化合物分子中与碳原子连接的氢被氟取代的一类元素有机化合物。分子中全部碳-氢键都转化为碳-氟键的化合物称全氟有机化合物,部分取代的称单氟或多氟有机化合物。由于氟是电负性最大的元素,多氟有机化合物具有化学稳定性、表面活性和优良的耐温性能等特点。有机氟化合物分为以下几类:①含氟烷烃。氟利昂是氟化的甲烷和乙烷,也可以含氯或溴。这类化合物多数为气体或低沸点液体,不燃,化学稳定,耐热,低毒。主要用作制冷剂、喷雾剂等,最常用的是氟利昂-11(CFCl3)和氟利昂-12(CF2Cl2)。这类化合物也是重要的含氟化工原料或溶剂。如二氟氯甲烷用于合成四氟乙烯;1,1,2-三氟三氯乙烷用于合成三氟氯乙烯,也是优良的溶剂。含氟碘代烷如三氟碘甲烷等为重要的合成中间体。一些低分子含氟烷烃和含氟醚具有麻醉作用,并有不燃、低毒的优点,可用作吸入麻醉剂,例如1,1,1-三氟-2-氯-2-溴乙烷(俗称氟烷)已广泛用于临床。②含氟烯烃。以四氟乙烯、偏氟乙烯和三氟氯乙烯等为代表。四氟乙烯为最主要的含氟单体,可以聚合成聚四氟乙烯,或与其他单体共聚合成多种含氟高分子。偏氟乙烯CF2=CH2在空气中的浓度在5.8%~20.3%之间时,遇火可爆炸,主要用于与其他单体共聚合制取含氟弹性体。三氟氯乙烯主要作为单体,用于合成均聚物或共聚物。③含氟芳烃。苯分子中的氢可以通过间接方法部分或全部用氟取代。氟苯为含氟芳烃的代表。多氟苯或全氟苯易与亲核试剂发生取代反应。
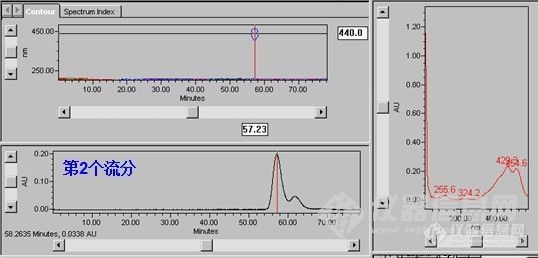
【发贴背景】昨天发的贴子“实验嗷嗷顺利,新化合物咣咣出(一)”,觉得标题不错,还蛮有意思的。私下我跟人说,这个月的极限体验贴,都要采用类似的标题,所以,现在“实验嗷嗷凄惨,化合物咣咣不纯(二)”出来了。上贴链接:http://bbs.instrument.com.cn/shtml/20111203/3687561/【实验过程】对某西红花苷类样品进行PHPLC,期望得到1-2个单体化合物。我曾经在一个贴子中说过,此类化合物最大的特点,就是不太稳定,这个问题一直困扰着我,我相信,它也困扰着其他做这方面实验的同行,呵呵。总之,这次分离的结果不是很理想。曾经的贴子:http://bbs.instrument.com.cn/shtml/20111102/3621305/【仪器试剂】水:重蒸水 甲醇:色谱纯,天津大茂 液相:Waters 高效液相,Waters 996 DAD检测器,Waters Model 600 controller液相色谱【液相色谱条件】 色谱柱:Ultimate XB-C18柱(5μm, 4.6x250mm)流动相:保密流速:1.0ml/min检测波长:全波长扫描【实验内容】1. 总样(制备前)分析图谱(略),原因:总样一般比较杂,是用其它品牌色谱柱分析的,极限的柱子,得多多爱惜,只用来分析纯化后的样品,这一点,上贴已经强调过。2. 制备液相时的色谱图(手机拍摄)从图中可以看出,流分1和流分2,以及它们与其它杂质的分离度是非常好的。所以,在进行制备的时候,进样量也是相当地大,单次进样量达到3.5ml。流分1是有可能得到一个单体化合物的,尽管他右侧的小包未能完全分离开来(经验)。流分2,从理论上来说,他是肯定能成为一个单体化合物的。http://ng1.17img.cn/bbsfiles/images/2011/12/201112041950_335461_2424913_3.jpg3. 各流分液相分析图谱流分1的色谱图,对于它,我早有心理准备,不纯也是正常的,毕竟右边还有那么大个没分开的小包。分析结果显示一个主峰和两个小峰,如果有Ultimate的半制备柱或者制备柱就好了,因为显示分离度挺好的。下次还得继续纯化,唉。http://ng1.17img.cn/bbsfiles/images/2011/12/201112042027_335474_2424913_3.jpg流分2的色谱图,居然不是单一峰形,这个非常让人失望。因为制备上面显示单一峰形,我以为能拿到至少一个单体化合物的,唉。这个样品,后续要继续纯化是非常困难的事,因为二者在50多分钟才出峰,如果要去进行制备液相将它们“拖开”,保守估计得2小时以上,而且进样量不会很大,凄惨啊!!http://ng1.17img.cn/bbsfiles/images/2011/12/201112042029_335475_2424913_3.jpg【小结与讨论】1. 这次实验很失败,当然了,人为的因素不大,这作为一个安慰的借口倒是蛮不错的,呵呵。2. 流分2继续纯化一下,是很有前途的;流分3继续纯化,是很凄惨的,很可能我就放弃了,因为这类化合物,不太可能出现新的化合物(个人经验)。3.流分1和流分2在制备的时候,其实分得挺开的,如果有跟制备柱类似的填料,我相信,用开放ODS就可以把它们分开了。可惜。。没有!!让老板买?别开这种玩笑,谢谢啊。4. 贴子里头讨论得差不多了,感觉很啰嗦。
我还记得,当年我拿到第一个化合物(是个结晶)。。。我兴奋,激动,我抑制不住内心的情感啊。。。我跑到老师办公室,尖叫:我分到化合物了!!!当时老师一把从椅子上跑过来。。。说:来来来!!放冰箱,小心变了!!!来,分享你的喜悦。。。
所谓高分子化合物,是指那些由众多原子或原子团主要以共价键结合而成的相对分子量在一万以上的化合物。 定义:由千百个原子彼此以共价键结合形成相对分子质量特别大、具有重复结构单元的有机[url=http://baike.baidu.com/view/63037.htm]化合物[/url]。 是由一类相对分子质量很高的分子聚集而成的化合物,也称为高分子、[url=http://baike.baidu.com/view/183139.htm]大分子[/url]等。一般把相对分子质量高于10000的分子称为高分子。高分子通常由103~105个原子以共价键连接而成。由于高分子多是由小分子通过聚合反应而制得的,因此也常被称为聚合物或[url=http://baike.baidu.com/view/328669.htm]高聚物[/url],用于聚合的小分子则被称为“单体”。

【发贴背景】上一篇极限体验中,我谈到了水层样品的分离,大家就此进行了“学术性非常弱,灌水性非常强”的讨论。其中,我跟美丽心情、小S版主还讨论到了“实验到底能不能用嗷嗷顺利、新化合物能不能用咣咣”这样的词来形容的问题,她们二位似乎对“嗷嗷”和“咣咣”这样的词汇比较敏感。虽然我们三方在讨论的过程中未达成一致,但此贴采用小S的建议,就叫“实验嗷嗷顺利,新化合物咣咣出”。 贴子链接(61楼):http://bbs.instrument.com.cn/shtml/20111130/3680419/index_7.shtml【实验过程】一句话就可以概括:样品Z进行制备液相,将收集到的各个流分进行分析。此贴侧重点在于分析过程,毕竟分析才是用的极限的柱子嘛。贴子标题一点也不吹牛,两个单体化合物,真的就是新化合物,目前正在进行2D-NMR测试(一维C、H谱已经测完了)。【仪器试剂】水:重蒸水 甲醇:色谱纯,天津大茂 液相:Waters 高效液相,Waters 996 DAD检测器,Waters Model 600 controller液相色谱【液相色谱条件】 色谱柱:Ultimate XB-C18柱(5μm, 4.6x250mm)流动相:保密内容流速:1.0ml/min检测波长:全波长扫描【实验内容】这一张是制备液相时拍的色谱图(电脑USB有问题,拷不下来,汗,将就看吧),什么牌子的制备柱就不用管了,这个不是重点。第1个流分是前面“一片峰”,重点是在第2、第3、第4个流分。结果分析之后,第2个流分居然不纯,只有第3、第4个流分显示单峰峰形,初步确定为单体化合物。http://ng1.17img.cn/bbsfiles/images/2011/12/201112032054_335290_2424913_3.jpg由于第1、第2个流分不纯,就不上它们的色谱图了。上上第3、第4个流分的色谱图吧,看着舒心,呵呵。这是第3个流分的色谱图http://ng1.17img.cn/bbsfiles/images/2011/12/201112032056_335291_2424913_3.jpg这是第4个流分的色谱图,保留时间、紫外吸收都跟第3个流分极为相似。事实上,经过NMR测试以后,他们的波谱行为也基本一致,属于同类型化合物,结构几乎没有差别,目前具体结构没有确定(当然,确定了你也不感兴趣;你感兴趣我也不可能告诉你),还在进行2D-NMR测试。http://ng1.17img.cn/bbsfiles/images/2011/12/201112032057_335292_2424913_3.jpg【小结与讨论】1. 制备前的总样色谱图就不附了啊,因为那不是极限的柱子分析的。极限的柱子,现在只用来分析一些纯化后的样品。柱子宝贵,得爱惜点啊。2. 制备那张图上反光的地方,是实验室的日光灯,特此声明,汗。3. 两个化合物,而且是。。。哦耶。4. 第1个流分已经放弃了,峰太多,而且量不多;第2个流分目前正在考虑要不要进一步纯化,量也不是很大(量太少的话,就算制备纯了,也没有办法进行NMR测试)5. Z-4有一点点小杂质,图上可以看到,不过不影响。6. 其它,欢迎讨论,或者提问。

维权声明:本文为chuanxinlian原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。浅谈甾体类化合物的分离与结构鉴定 在植物环己烷或石油醚萃取部分,有很多小极性的化合物,并且这些化合物之间的极性差异也很小,分离时可能会遇到很多困难,即使分离得到了单体化合物,结构解析时也是一件很不容易的事,不管是氢谱还是碳谱中,都在高场区出来一堆信号,有时氢谱中的积分都不一定能积准,碳谱的高场区就像个小树林,密密麻麻的全是信号,看着就头疼。由于我曾从一植物的环己烷层中分离得到了多个甾体类化合物(主要为豆甾类),下面就我在实验过程中在分离和解析甾体类化合物的一点经验跟大家分享一下。1.甾体类化合物的分离纯化 由于我做的这一部分的成分极性很小,其中的甾体类化合物均不连糖,并且我所得到的化合物的结构很相似,有的只相差一个双键或只差一个羟基或羰基,总之,结构类似,极性也就相差不大,分离也就不容易了。由于极性小,我采用的分离手段只能局限在硅胶柱或板、凝胶柱(Sephadex LH-20)。首先把粗浸膏过一遍大的硅胶柱,已达到按照极性大致砍一下段的效果,由于环己烷萃取部分有很多色素,所以接下来采用过Sephadex LH-20柱将其中的色素除去,最后得到多种没有色素的流分,这里面的成分也是很杂的,有多种类型的化合物,如黄酮类、二萜内酯类、甾体类等,由于各类型化合物间的分子量有很大的差异,接下来就可以过一个又细又长的Sephadex LH-20柱,流速一定不能太快,并且流分要接的很细,我们一般用5ml的小瓶接,尽量避免不必要的交叉。这样就有可能将甾体类化合物与其他类型的化合物分开了,最后一步,在硅胶薄层板上看一下,如果成分简单,分离度良好,就采用制备薄层色谱,将单体化合物得到。 需要指出的是,制备薄层时,由于很多甾体类化合物在紫外灯下没有任何紫外吸收,这时千万不要认为没有紫外吸收的地方就没有化合物。比如这块板上的: http://ng1.17img.cn/bbsfiles/images/2010/10/201010220007_252894_1745326_3.jpg 此时,一定要在点小硅胶板时用通用显色剂显色,确定哪里有点,该流分总共有几个点,各个点之间的上下关系,有没有紫外吸收之类的,分析清楚了,最后再去做制备薄层,如果有没有紫外吸收的点,在刮板前可以先用玻璃或报纸将板的大部分挡住,在板的另一边显色,找到该点的具体位置,以免漏掉重要的点。2.豆甾类化合物的结构解析 因为我分到的甾体类化合物主要为豆甾类,所以我就只介绍此类化合物的结构解析了。下面是我在解析过程中总结的该类化合物的谱学上的特点:2.1 氢谱中最高场的信号为18-CH3,化学位移一般在0.7ppm左右。2.2 碳谱中最高场的信号同样为18-CH3,化学位移一般在12.0ppm左右。实例列举:http://ng1.17img.cn/bbsfiles/images/2010/10/201010220010_252897_1745326_3.jpg1H-NMRhttp://ng1.17img.cn/bbsfiles/images/2010/10/201010220011_252898_1745326_3.jpg13C-NMRhttp://ng1.17img.cn/bbsfiles/images/2010/10/201010220011_252899_1745326_3.jpg2.3 甾体母核上的羟基的取向不同,与其所连的碳的化学位移也会有所不同,一般情况下,α-OH连接的碳化学位移一般小于70.0ppm,而β-OH连接的碳化学位移一般大于70.0ppm。举例如下:http://ng1.17img.cn/bbsfiles/images/2010/10/201010220011_252900_1745326_3.jpg2.4 如果你拿到一套新的谱图,看到高场区有很多聚集在一起的碳氢信号,并且氢碳谱中最高场的化学位移值分别在12.0和0.7ppm左右,在你不确定它什么类型的化合物的时候,可以考虑一下是不是甾体类化合物(三萜类最高场的碳信号在18ppm左右,基本上没有12ppm左右的碳信号)。最后,希望我的原创能给某些人起到抛砖引玉的作用。
[color=#333333]橄榄(Canarium album L.)为我国珍贵的药食两用资源,具有解酒护肝、抗菌消炎、抗病毒和解毒等药理功效,橄榄中酚类化合物是其主要的药效成分,但国内外有关橄榄酚类化合物组成的研究报道不多。本论文对我国橄榄果实中的酚类化合物进行提取、分离和纯化,并对酚类化合物单体的化学结构进行鉴定研究,以明确橄榄酚类的具体组成,对于橄榄资源的深加工利用和橄榄中药的药理研究具有重要的指导意义和应用价值。首先采用化学和仪器分析方法对福建闽侯檀香橄榄果实不同部分的化学组成进行了分析测定,[/color]
1、加聚反应制得的高分子化合物 加聚反应制得的高分子化合物,其命名习惯上是在原料名称之前,加一个“聚”字。如,氯乙烯的聚合物,称为聚氯乙烯;四氟乙烯的聚合物,称为聚四氟乙烯;甲基丙烯酸甲酯的聚合物,称为聚甲基丙烯酸甲酯。 2、缩聚反应制得的高分子化合物 缩聚反应制得的高分子化合物,其命名习惯上是在原料名称之后,加“树脂”二字。如,酚醛树脂、环氧树脂、脲醛树脂等。事实上,加聚产物在未制成成品之前也常以“树脂”称之。如,聚乙烯树脂、聚丙烯树脂等。 3、聚酰胺类高分子化合物 聚酰胺类高分子化合物,其命名是在聚酰胺后面加上数字,该数字表示单体中碳原子的个数。例如,由己二胺和己二酸缩聚而成的高分子化合物,称为聚酰胺-66;由癸二胺和癸二酸缩聚而成的高分子化合物,称为聚酰胺-1010。 4、合成橡胶类高分子化合物 合成橡胶类高分子化合物,其命名是在橡胶二字的前面加上能代表单体名称的几个字。如1,3-丁二烯与苯乙烯的聚合物称为丁苯橡胶;2-氯-1,3-丁二烯的聚合物称为氯丁橡胶;1,3-丁二烯与丙烯腈的聚合物称为丁腈橡胶;异戊二烯的聚合物称为异戊橡胶,依此类推。 5、商品名称 商业上为了方便,常给某些合成纤维以商品名称,称为“某纶”。 (1)锦纶(或尼龙)聚酰胺类合成纤维,它的商品名称叫“锦纶”或“尼龙”,如,锦纶-6、锦纶-66,尼龙-610等。 凡是后面有两个或两个以上数字的,表示这种聚酰胺纤维是由二元胺和二元酸两种单体缩聚而成的。前面的数字是二元胺的碳原子数,后面的数字是二元酸的碳原子数。如,尼龙-610是由己二胺和癸二酸缩聚而成的。 凡是后面只有一个数字的,表示这种聚酰胺纤维是由某碳原子个数的内酰胺聚合而成的。如,锦纶-6是由己内酰胺聚合而成的。 (2)涤纶 聚酯纤维是指纤维分子中各个链节,都是以酯基相连接形成的高分子化合物,商品名称叫“涤纶”。目前,工业生产中产量最大的涤纶是聚对苯二甲酸乙二酯,俗称“的确良”。 另外,还有一些常见的高分子化合物的商品名称,如,“腈纶”、“丙纶”、“氯纶”、“维尼纶”,等等。 “腈纶”——聚丙烯腈纤维; “丙纶”——聚丙烯纤维; “氯纶”——聚氯乙烯纤维; “维尼纶” ——聚乙烯醇缩甲醛纤维。

(1)-3-7 分子式索引 (Formula Index, FI)分子式索引于 1920 年第 16 卷起推出. 报导的内容简洁, 只提供化合物的名字、登记号与文摘号三种信息 (下图), 是辅助索引. 需要进一步了解化合物的详细信息则需要查找化学物质索引. 使用分子式索引的时机包括不清楚化合物的命名, 以及快速了解某化合物是否有人研究过.CA 对 分子式的编排采用简单易记的 Hill 系统, 而不用 20 世纪初盛行的 Richter 系统 (Richter 系统字母次序为 C, H, O, N, Cl, Br, I, F, S, P 等, 例如 C10H5ON4BrS). Hill 系统以 C, H 为主体, 然后其它原子按照英文字母顺序排列, 例如 C10H5BrN4OS. 如果化合物没有 C, 则全部按照字母次序, 例如氢溴酸为 BrH. 想进一步了解 Hill 系统可以查阅 J. Am. Chem. Soc., 1900, 22 (8), 478-494.有些结构不明确的化合物在分子式索引中暂不命名, 但提供沸点、熔点、质谱等理化数据. 对于高聚物或有机盐 (如醇、酚、胺的金属盐) 的查阅从化合物的母体着手, 例如高聚物从单体; 季铵盐 (C6H5CH2)(CH3)2(CH3CH2)N+ I- 从C11H18N; 乙醇钠 C2H5ONa 从 C2H6O 母体着手.分子式索引的编排、阅读与查找范例如下:http://ng1.17img.cn/bbsfiles/images/2011/10/201110190538_324733_1631320_3.jpg练题 1: 查 1997 年 126 卷报导下列化合物 (分子式 C19H20ClN3S) 的文摘Piperidine 1--4--1H-pyrazol-3-yl 答案 1: 126: P31349v练题 2: 查 1992-1996 期间报导 dichlorodiiodomethane (CCl2I2) 的文摘号答案 2: 117: 14730j; 120: 63571d; 120: 106321s[/fo
维权声明:本文为环烯醚萜原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。环烯醚萜类化合物分离纯化心得体会基本介绍 环烯醚萜(iridoids)为臭蚁二醛(iridodial)的缩醛衍生物。臭蚁二醛是由伊蚊(Iridomyrmex detectus)的防收性分泌物中分得的物质。自1958年的Halpem和Schmid确定的环烯醚萜的基本骨架以来,各国学者对该类化合物作了大量深入的研究。环烯醚萜类化合物具有多种生物活性,近来受到极大关注,发展也很迅速。 环烯醚萜类主要分为:环烯醚萜类、裂环环烯醚萜类、3,4-位无取代的环烯醚萜类、聚合环烯醚萜类等等。本人做的是普通类的环烯醚萜类化合物,且以其苷居多,做的比较浅,下面斗胆一谈,各位看官莫要见笑。提取部分 环烯醚萜苷类化合物在醇(甲醇、乙醇)中溶解度较好,部分苷类在水中溶解度也很好。本人在对某植物进行提取的时候,实际上并非针对这类化合物,采用的是60%的乙醇/水,是为了兼顾各类成分。后来在实验过程中发现,60%的乙醇/水条件下,这类化合物的提取率是很高的。 注释:其实如果要针对性分离,可以将提取液简单处理后进行D101大孔柱色谱,对环烯醚萜类化合物进行富集。萃取部分 提取之后,将药液进行浓缩,至无醇味混悬于水中,然后进行萃取。萃取的过程为:等体积的环己烷、乙酸乙酯、正丁醇分别萃取三次,合并各层提取液浓缩得各层浸膏。就目前实验进展情况来看,环烯醚萜苷类化合物主要集中在正丁醇层,水层也有一部分(我目前还没开始这部分工作)。 注释1:萃取的过程,涉及到溶剂的回收,由于这类化合物在高温下不太稳定,所以用旋转蒸发仪进行减压回收溶剂的时候,温度不能过高,我采用的温度是60度(其实60度已经很高了,但是没办法,不设60度,正丁醇回收不了)。 硅胶柱色谱 正丁醇层进行硅胶柱色谱分离,采用氯仿/甲醇梯度洗脱,样品500g,拌样硅胶1500g,柱床硅胶500g,洗脱梯度为50:1→20:1→10:1→5:1→2:1→1:1→0:1。实验的过程中发现,环烯醚萜类化合物,主要集中在氯仿:甲醇=10:1和5:1部分。 注释1:选择硅胶柱色谱,也是人之常情,此处也可选择D101大孔柱色谱,对这类化合物进行富集; 注释2:选择氯仿/甲醇系统,是因为经过小试,此系统对样品分离较好,最重要的,样品在此系统中成点性很好; 注释3:拌样1500g,柱床500g,你没有看错,我没有写错。书上说,拌样:柱床在1:1-1:10甚至1:20或者1:50,而我这里却是3:1。我可以很负责任地告诉你,没有必要按书上的说法,柱床500g足够了,分离效果一点也不差。以前做另外一个植物,拌样用了3000g,柱床才600g,分离效果也不差,一点问题都没有; 注释4:梯度的选择,建议6-8个。梯度太少,各流分成分可能过于复杂;梯度太多,后续分离麻烦。这种大型硅胶柱色谱,属于平常所说的“粗分”,不宜太多,不宜太少,不然就是给自己找麻烦。http://ng1.17img.cn/bbsfiles/images/2010/10/201010061143_249374_1745326_3.jpgODS柱色谱 包括开放型ODS柱色谱和中低压型ODS柱色谱,其实原理一样,只是规模大小不同而已。 我将5:1洗脱的样品进行中低压ODS柱色谱,采用甲醇/水梯度洗脱,水→10%甲醇/水→20%甲醇/水→30%甲醇/水→50%甲醇/水→甲醇,实验结果表明,ODS柱色谱对此类化合物具有良好的分离效果。 注释1:环烯醚萜苷类化合物一般极性较大,一般集中在10%甲醇/水、20%甲醇/水、30%甲醇/水部分; 注释2:水洗下来的,一般为糖苷类化合物,我从此流分中分离得到几个糖类化合物(题外话); 注释3:ODS柱色谱可以多次进行,反复纯化,利用ODS柱色谱可以得到部分单体化合物;http://ng1.17img.cn/bbsfiles/images/2010/10/201010061144_249375_1745326_3.jpg(注:此图为未合并相同流分前的点板情况)制备液相(反相) 从ODS柱色谱上洗脱的样品,经过分析,可以考虑进行制备液相,半制备液相等等。 事实上,一般而言,PHPLC也是获得环烯醚萜类单体化合物最重要的手段之一。 流动相可采用甲醇/水,若峰形不好,可加入少量乙酸改善(这一招屡试不爽)。 波长的选择,可以使用230nm、240nm都可以(我们有PAD检测器,验证过)。其它一些化合物,由于连上芥子酰基,对羟基香豆酰基,还可以选择320nm的吸收。 色谱柱一般是C18的居多(目前未使用过其它柱子),品牌好像都还行(我们主要使用YMC)。凝胶其它填料 由于分离原理的缘故,使用凝胶对环烯醚萜这一类化合物(分子量相差不大,且结构极为类似)进行分离,前景似乎并不明朗。但是,用凝胶将这一类化合物与其他类型的化合物分离,效果还是很不错的。 其它如大孔树脂,前面提过,用来富集是个不错的选择;聚酰胺,我没有使用过,不过从其分离原理来看,对这类化合物不会很敏感。显色剂的选择 部分环烯醚萜类在254nm紫外下有暗斑,这个很实用; 最常用的是浓硫酸-香草醛显色剂:母核上有羟基取代的显蓝色;母核上无羟基取代显紫红色(非绝对); 其它显色剂如硫酸乙醇、碘等等都可以,我个人偏爱浓-香显色剂。结构测定与解析(简) 环烯醚萜类化合物进行NMR测试,首先氘代甲醇,这个没有任何疑问。 我在一些文献中,也看到一些特例,比如使用重水、氘代DMSO,这些基本可以忽略。 关于结构解析部分,此处不再赘述。
氨基十二酸,一种长碳链的的两端分别带有氨基和羧基的化合物,是合成尼龙12的一种单体。我现在想用液相的方法测定,首先该化合物不溶于一般的有机溶剂,只能溶于乙酸(溶解的具体原理不详),因此只能先用乙酸溶解之后再用乙腈溶解,然后进样测定。色谱条件:色谱柱:C8波长:210nm流速:1ml/min流动相用5%的乙腈跑,只有一个溶剂峰,应该是乙酸的溶剂峰,流动相改用100%的乙腈仍然只有一个峰,后来用甲醇:正己烷=3:1的流动相洗脱,仍然只有一个峰,请问这是什么问题?是氨基十二酸留在柱子上洗脱不下来吗?还是氨基十二酸跟溶剂峰一起出来了?希望有经验的大神能够指导一下,我应该怎么办?或者提供一下类似于该化合物结构的研究资料,我研究一下。
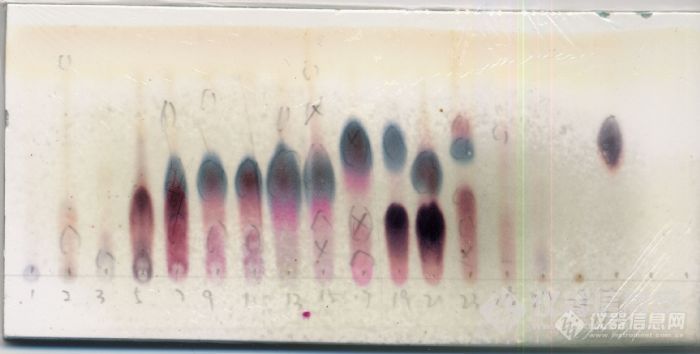
维权声明:本文为xiaodu2007693原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。黄酮类化合物的分离体会 【黄酮类化合物简介】 黄酮类化合物(flavonoids)是一类存在于自然界的、具有2-苯基色原酮(flavone)结构的化合物。它们分子中有一个酮式羰基,第一位上的氧原子具碱性,能与强酸成盐,其羟基衍生物多具黄色,故又称黄碱素或黄酮。黄酮类化合物在植物体中通常与糖结合成苷类,小部分以游离态(苷元)的形式存在。绝大多数植物体内都含有黄酮类化合物,它在植物的生长、发育、开花、结果以及抗菌防病等方起着重要的作用。【本人研究内容】 我做的是某中药的化学成分研究,该中药中主要为黄酮类成分,遵循传统用药原则,我采取水煎煮,之后水提液浓缩至一定体积,过大孔吸附树脂,乙醇水梯度洗脱,梯度:水、20%乙醇、40%乙醇、60%乙醇、80%乙醇。每个梯度做为一个流分,再进行细分。下一步主要是运用聚酰胺柱色谱,中低压ODS,Sephadex LH20进一步分离,最后制备液相得到单体化合物。【经验分享】 主要和大家分享我使用聚酰胺柱的经验,我一般是聚酰胺薄膜结合聚酰胺柱,类似于硅胶板结合硅胶柱。所用的聚酰胺为30-60目,聚酰胺薄膜一盒50片,规格为8cm x 8cm,70块钱左右,可以根据需要剪成条状使用。聚酰胺薄膜常用的溶剂系统说明书上有,我常用甲醇-水,因为黄酮类化合物有一定的酸性,展开剂要加点酸,甲酸乙酸都可以,抑制其解离,保持游离状态的话,样品成点性好,否则拖尾很严重。在此需要指出的是,聚酰胺可以用反相溶剂系统,如甲醇水;也可以用正相溶剂系统,如二氯甲烷甲醇。要根据自己样品的极性来定。以我的80%乙醇洗脱物为例,进行详细的叙述。此部分极性较小。我用二氯甲烷溶解,拌样,挥干。洗脱系统为甲醇水,聚酰胺柱起始用水装柱,上样,上面要放一块棉花,然后用玻璃塞子压住(聚酰胺比较轻,防止加溶剂时冲起来),甲醇水梯度洗脱。之前点板显示,50%甲醇水刚刚展开一点,因此起始用50%甲醇水洗脱,然后梯度设为55%、60%、65%、70%、75%、80%、85%、90%、最后甲醇冲柱。过程中每100mL一接,蒸干转移至小瓶。最后用一大块聚酰胺对得到的流分进行合并。得到大概9-10个流分,HPLC-DAD分析,找到合适的液相色谱条件之后,用制备液相进行制备。【部分聚酰胺薄层谱图展示】http://ng1.17img.cn/bbsfiles/images/2010/09/201009211940_245940_2160661_3.jpg http://ng1.17img.cn/bbsfiles/images/2010/09/201009211940_245941_2160661_3.jpg左图:展开剂:(甲醇:水90:10)+2滴乙酸; 右图:展开剂:(甲醇:水80:10)+2滴乙酸,最右边的点为总样显色剂为:三氯化铁乙醇溶液【体会和教训】 黄酮类化合物用聚酰胺分离效果非常好,但是死吸附较严重,样品比较多时可以使用,少就算了。凝胶Sephadex LH20效果也非常好,建议多使用。黄酮类成分溶解性比较折磨人,但提醒一点在进行制备液相时,样品一定要用流动相溶解,或接近流动相,多加点总会溶解的,然后用0.45μm滤膜过滤。不用流动相溶解很容易把柱子弄裂分。原因在于,进样之后,样品析出,堵了柱塞板,导致局部流速过快,冲塌固定相。我就把老师的预柱整裂分了,多亏了预柱啊,保护了柱子,要不然老师肯定疯了,但是后来寄出去修预柱,别人检测又很神奇地自动好了,这有点解释不了,呵呵,这事也不是常能碰到的,最好按规矩来,否则耽误别人实验不太好。先就写这么多,实验也不是那么成功,但拿出来和大家交流,不对的地方请批评指正。【样品导致预柱裂分的后果-色谱图展示】http://ng1.17img.cn/bbsfiles/images/2010/09/201009212004_245944_2160661_3.jpg http://ng1.17img.cn/bbsfiles/images/2010/09/201009212007_245946_2160661_3.jpg左图为:样品制备时色谱图,当时已裂分右图为:预柱用纯品检测色谱图,连续进样2针,可见预柱裂分严重。
是不是所有的化合物都能在TIC图上显示?比如4个化合物应该有4个色谱峰?如果只看见2个峰是咋回事呢?
[size=4]我们想买甲硫醇,甲硫醚,乙硫醇,乙硫醚,二甲二硫,一甲基乙基二硫等硫醇和硫醚类的化合物的液体纯化合物,大家知道哪里有的买吗?如果知道的话,麻烦告诉我一下啊,谢谢了![/size]

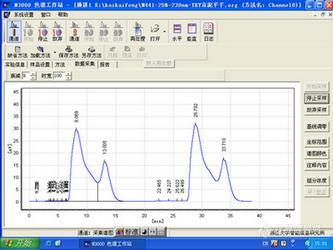
【背景介绍】喹宁酸类化合物具有多种具有多种生物活性,如抗组胺、抗氧化、肝保护、抗菌等。而且,其单取代、双取代或三取代化合物在体内化外都显示出良好的抗HIV病毒活性,具有很好的开发前景。【喹定酸类化合物结构】二取代类喹宁酸类化合物是指在R1和R2、R1和R3、或者R2和R3两个位置均有取代的化合物。取代基可能为咖啡酰基(A)、芥子酰基(B)、阿魏酰基(C)等基团的自由组合,即R1、R2、R3可能为A、B、C的排列组合,R4可能为氢也可能为甲基或者乙基等基团。由于R1、R2、R3所处取代位置的化学环境相似,所以在结构鉴定过程中,取代位置容易混淆。http://ng1.17img.cn/bbsfiles/images/2011/10/201110042032_321102_1745326_3.jpg【文章内容】本文通过自己制备的部分单体化合物,利用此类化合物在液相中保留时间的不同,对某植物中存在的部分喹宁酸类化合物(10个:G1~G9)进行指认(涉及保密,本文只透露2个化合物(G4、G7)的相关内容)。对于明确此植物的化学成为具有重要的指导意义。【仪器试剂】水:重蒸水甲醇:色谱纯,天津大茂 乙酸:分析纯,天津大茂 HPLC:Waters 高效液相Waters 996 DAD检测器Waters Model 600 controller液相色谱【色谱条件】 色谱柱:Ultimate XB-C18柱(5μm, 4.6x250mm)流动相:60%甲醇/水(V:V),水含0.01%乙酸流速:1.0ml/min柱温:常温(约25度);检测波长:全波长检测;进样量:10微升;【样品制备】涉及保密,此处略去,见谅。重点在色谱行为。【色谱行为】 某植物样品——总样分析色谱图http://ng1.17img.cn/bbsfiles/images/2011/10/201110062154_321549_1745326_3.jpg指认的G4峰:3,4-二咖啡酰基喹宁酸甲酯http://ng1.17img.cn/bbsfiles/images/2011/10/201110062155_321550_1745326_3.jpg指认的G7峰:3-咖啡酰基-5-芥子酰基喹宁酸甲酯http://ng1.17img.cn/bbsfiles/images/2011/10/201110062156_321551_1745326_3.jpg【结果与讨论】1. 本实验对某植物中所含的喹宁酸类化合物进行了指认(约10个),明确了该植物中含有的化学成分,为以后工作的开展奠定了一定的基础。同时,从色谱图中可见,还有部分化合物由于量小未进行指认,这部分工作将是以后工作的重点;2. G4和G7的色谱图虽然可见一些含量很少的小杂质,但并不影响此次实验分析。由于双取代基团的存在,使得它们的紫外吸收峰并不“干脆”,如果是单取代吸收,将在230nm和320nm处显示两个“干脆尖锐”的紫外吸收峰。这一点,有利于识别单取代和双取代喹宁酸类化合物。3. 另外,不得不反复提出,在进样量不同的时候,通过DAD检测器得出的紫外吸收值有所变化。这提示我们,DAD检测器得出的紫外吸收只应该做为一个参考值,需要引起注意。4. 总样分析色谱图中,由于出峰时间过短及流动相等条件摸索不到位,部分峰形没有完全分离,此部分工作将继续进行。尽管如此,Ultimate XB-C18柱在分析的过程中,表现出了分离度好、柱效高、柱压稳定等优秀性能,让人满意。后续开展的工作,如果涉及指纹图谱类似的相关实验内容,Ultimate XB-C18柱将成为种子选手,绝对首发。
请问各位大侠,想要测气体中的成分需要用什么设备?想知道气体中都有哪些化合物!
请问五元环的手性化合物 顺反异构体作检测时至少作哪几种NMR普能够判断出来?谢谢
请教大家:我现在在做一个化合物,是枸橼酸钾、枸橼酸钠通过特殊的化学反应生成一种离子化合物。但是现在有个问题,两者反应后不可能全部生成化合物,肯定还有没有反应的物质,如果生成率小于80%那不能满足我的要求了。我现在想知道的是里面有多少“枸橼酸钾钠”化合物,还有他们的分子构成是什么,该用什么方法分析呢?
一、Markes1、Tenax:芳香烃化合物(苯除外),非极性化合物沸点100°C,极性化合物沸点150°C2、热脱附仪:该热脱附仪符合的关键方法有US EPA TO-17总结:Markes设备只能是对气体物质中的有机化合物进行检测,无法检测固体物质、液体物质和无机气体。n-C7 to n-C30,沸点100°C to 450°C。芳香烃化合物(苯除外),非极性化合物沸点100°C,极性化合物沸点150°C,二、Finnigan1、毛细管柱: 产品简介:来自西班牙TK公司的色谱柱TRB-MetaX5 30M*025MM*025UM2、该仪器为离子源、四极杆。我们单位仪器由以上两个公司的组成,Markes公司和Finnigan公司。因此我归纳我们单位可做的样品应有如下特点:1、该仪器只能对气体物质中的有机化合物进行检测;2、该有机化合物为非极性化合物;3、n-C7 to n-C30;4、沸点100°C to 300°C。现在领导要求我把具体的化合物罗列出来。我可闷掉了,请求大家帮我罗列一下。谢谢各位了!
极性化合物完美分离保留 Atlantis色谱柱 给极性化合物和非极性化合物的保留提供了完美的平衡下载Atlantis色谱柱介绍资料(PDF) Atlantis 色谱柱使用高纯度硅胶及双键键合 C18 技术,并对填料的孔径大小、端基封口以及 C18 的键合密度进行优化,从而使 Atlantis 色谱柱具有: 对极性化合物保留能力强,在水流动相中性能稳定,低 pH 条件下色谱柱寿命长,与质谱兼容,色谱峰形优异,重现性好等优点。Atlantis 色谱柱目前提供3um和5um两种粒度填料,键合相类型则有dC18和HILIC(亲水交互作用)两种类型。Atlantis 资料目录:极性化合物的保留ATLANTI dC18 反相HPLC分析的理想选择极性化合物的保留增强色谱柱的长寿命和低pH稳定性为使用水溶液流动相优化的填料即使没有内嵌极性官能团也与水溶液兼容完全端基封口色谱柱的优势极性和非极性化合物保留的最佳平衡ATLANTI dC18 色谱柱在肽谱方面的应用ATLANTI dC18快速分析柱优异的重现性轻松放大、更长、可预计的色谱柱寿命保留和高上样量的优化亲水相互作用色谱柱(HILIC)ATLANTIS HILIC 硅胶色谱柱适合于在反相色谱柱上不能保留的化合物的分离HILIC能够提高ESI-MS的灵敏度简化样品制备过程Atlantis dC18 色谱柱使用过程中的问题解决方案与故障排除HILIC Silica 色谱柱HILIC与反相色谱互补的选择性提高LC/MS灵敏度世界一流的宽PH值 极限色谱柱 Waters XTerra色谱柱 PH1-12 无与伦比的批间重现性 极佳的对称性 纯水流动相 Symmetry色谱柱100%纯水流动相 宽PH值 XTerra RP纯水极限色谱柱 PH2-12SunFire色谱柱 最好的低PH值稳定性 最佳粒度和批次重现性 满足制药行业最严格的性能要求
测定液体化合物中水含量的方法
请问一下,我们ICP测无机化合物和有机化合物,何为无机化合物,何为有机化合物。能否具体举实例来说明一下呢?
手性柱能分开手性化合物,能分开同分异构体吗?比如二甲苯3种同分异构体。
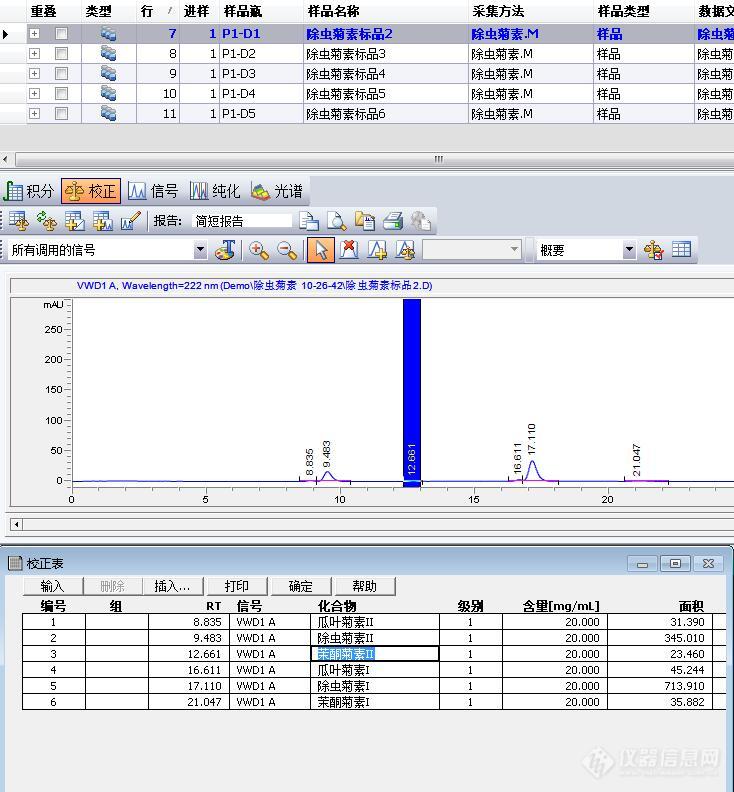
[align=center][size=18px]峰面积加和高阶用法之化合物组[/size][/align][align=left]在分析检测中,常常会有需要对多个化合物做汇总计算的情况。通常可以使用峰面积加和的方式计算。但对于多个化合物,化合物之间有其他峰的情况下,不能使用加和方式。此时可以使用化合物组来实现。下面就以除虫菊素为例,该产品包含6个化合物,分别是除虫菊素I,除虫菊素II,瓜叶菊素I,瓜叶菊素II,茉酮菊素I,茉酮菊素II,标品上只标注了6个化合物浓度总和,最终产品检测结果也只需要计算总浓度。[/align][align=left]1. 新建校正表,输入化合物浓度,这里输入总浓度20,并输入各个化合物名称[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301281424_6598_2963297_3.jpg[/img][/align][align=left]图1 建立校正表,输入浓度[/align][align=left][/align][align=left]2. 在校正表的“组”里输入组编号,本例中6个化合物编号都为1,然后在弹出的对话框里输入组名称“除虫菊素”,并勾选“组含量计算”,如图[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301280849_2653_2963297_3.jpg[/img][/align][align=left]图2 设置组编号及组名[/align][align=left][/align] 3. 继续添加新的级别,添加完成后,点击菜单栏校正/化合物组[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301285153_7838_2963297_3.jpg[/img]图3 打开化合物组4. 在化合物组细节中,在”组校正设置”中输入另外4个级别的浓度[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301283026_5851_2963297_3.jpg[/img]图4 输入各个级别的浓度5. 输入浓度后点确定,此时可以看到各个化合物按照面积百分比方式计算得到各自的浓度,并绘制标准曲线。[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301287402_7183_2963297_3.jpg[/img]图5 校正曲线(部分)6. 再设置报告格式为外标法,预览报告,即可得到样品各个化合物的浓度以及最后浓度加和[img]https://ng1.17img.cn/bbsfiles/images/2023/07/202307011301288534_1038_2963297_3.jpg[/img]图6 最终报告总结:此方法类似于MassHunter中的面积加和,只知道化合物总浓度,然后将面积加和后,根据峰面积百分比来计算各个化合物浓度,最后计算得总浓度。是面积加和的变种。
有人做过这方面的统计吗?有机化合物一旦进入人体很难代谢出去,那么我们经常接触的有机物呢?比如,苯、甲苯、酯类、卤化物等。我们做检验会经常用到这些物质,危害有多大呢?[em09511]
各位老师: 实验中碰到下面难题:一个含有2个手性中心的化合物,有4个对映异构体;顺利将该化合物四个对映体拆分后,利用在线旋光测出了这四个对映体的旋光性,依次为 (+)/(-)(+)/(-);问题是:这四个对映体的绝对构型(R)/(S)目前条件没有办法得到;在如何描述这个对映体上碰到了麻烦! 对于一个仅含有一个手性中心的化合物A,即便不知道其绝对构型,如果测定出了其对映体的旋光性,可以用 (+)-A 或 (-)-A 来表示该手性化合物的两个对映异构体;但对与含2个手性中心,有4个对映体的化合物,不知道其绝对构型,仅知道旋光性,按照上面的命名显然会造成歧义;目前也没有查到率属于上述情况的能供参考的文献,请各位老师支招,有没有比较好的办法来表示这四个对映体?麻烦了,非常感谢!
自19世纪末以来,稀有气体元素不能生成热力学稳定化合物的结论给科学家人为地划定了一个禁区,致使绝大多数化学家不愿再涉猎这一被认为是荒凉贫瘠的不毛之地,关于稀有气体化学性质的研究被忽略了。尽管如此,仍有少数化学家试图合成稀有气体化合物。1932年,前苏联的阿因托波夫(A.R.Antropoff)曾报道,他在液体空气冷却器内,用放电法使氪与氯、溴反应,制得了较氯易挥发的暗红色物质,并认为是氪的卤化物。但当有人采用他的方法重复实验时却未获成功。阿因托波夫就此否定了自己的报道,认为所谓氪的卤化物实际上是氧化氮和卤化氢,并非氪的卤化物。1933年,美国著名化学家鲍林(L.Pauling)通过对离子半径的计算,曾预言可以制得六氟化氙(XeF6)、六氟化氪(KrF6)、氙酸及其盐。扬斯特(D.M.Younst)受阿因托波夫的第一个报道和鲍林预言的启发,用紫外线照射和放电法试图合成氟化氙和氯化氙,均未成功。他在放电法合成氟化氙的实验中将氟和氙按一定比例混合后,在铜电极间施以30000伏的电压,进行火花放电,但未能检验出氟化氙的生成。扬斯特由于对传统观念心有余悸,没有坚持继续进行实验,使一个极有希望的方法半途而废。一系列的失败,致使在以后的30多年中很少有人再涉足这一领域。令人遗憾的是,到了1961年,鲍林也否定了自己原来的预言,认为“氙在化学上是完全不反应的,它无论如何都不能生成通常含有共价键或离子键化合物的能力”。







 我要推广仪器
我要推广仪器
 下载APP
下载APP