我们的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]源是ESI源,它的应用范围很广,能测定小分子的化合物,也能测定蛋白质等大分子化合物,与三重四级杆链接,能不能测大分子化合物呀,我们的仪器是安捷伦的6460,这台设备的扫描范围是5到3000,是不是就没法测定大分子化合物?
请教大分子化合物首选什么柱子?
大分子化合物首选什么柱子?
所谓高分子化合物,是指那些由众多原子或原子团主要以共价键结合而成的相对分子量在一万以上的化合物。 定义:由千百个原子彼此以共价键结合形成相对分子质量特别大、具有重复结构单元的有机[url=http://baike.baidu.com/view/63037.htm]化合物[/url]。 是由一类相对分子质量很高的分子聚集而成的化合物,也称为高分子、[url=http://baike.baidu.com/view/183139.htm]大分子[/url]等。一般把相对分子质量高于10000的分子称为高分子。高分子通常由103~105个原子以共价键连接而成。由于高分子多是由小分子通过聚合反应而制得的,因此也常被称为聚合物或[url=http://baike.baidu.com/view/328669.htm]高聚物[/url],用于聚合的小分子则被称为“单体”。
具一些文献说,一些大分子(例如树枝状化合物)处于分子内部的一些氢核磁不太好扫出来,结果就是核磁积分变小,对于这个问题如何解决呢?
今天有个学生测试两个膜蛋白之间有无相互作用。目前测试分子相互作用方法技术有很多,但是很多都是很复杂和麻烦了,分子相互作用也是很热门的技术。但是有些时候可以比较简单的,采用动态光散射测试生物大分子粒径。这个很好理解,目前我们这台仪器是动态光散射,根据布朗运动,利用分子运动快慢判断其粒径大小。分子越小,布朗运动越快,分子越大,运动越慢。首先分别测试出A和B两个膜蛋白的粒径,然后将两者混合,再测试粒径。如果粒径是A+B的话说明这两个有相互作用。缺点:1:动态光散射只是能够简单定性判断一下,A和B有没有相互作用,不能够精确计算其相互作用的解离常数,可提供的数据参数少。2:不适合做小分子。其能够识别范围都是1nm以上的,小化合物分子无法测试3:对样品纯度和准确性要求较高,无法测试复杂混合样品优点:简单快速!!!与目前其他测试技术相比的明显优势,且基本没有额外的消耗成本。纯粹简单的光学原理,测试非常快,30min~60min就可以完全测试结束,而且技术简单易理解。
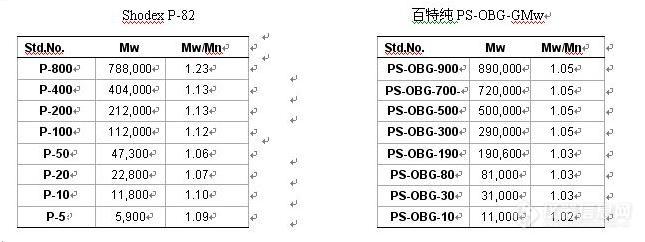
目前,高分子碳水化合物正被广泛应用于食品,啤酒,制药等行业,如香菇多糖,羧甲基纤维素,透明质酸,半乳甘露聚糖等,但对于不同分子量的碳水化合物来说,它们的作用都是不同的,因此对于准确测定高分子碳水化合物的分子量就显的尤为重要。 目前,绝大数公司,高校和科研院所都使用凝胶渗透色谱(GPC)法来测定高分子物质分子量,这种方法相对于光散射法来说具有操作简单,经济快速等优点,因此在各行业得到广泛的使用。对于GPC法来说,在测定待测样品时,需要选择标准品来做标准曲线,因此对于准确测定高分子碳水化合物的分子量来说,标准品的正确选择就显得尤为重要。Pullulan和Dextran是两种从微生物中提取的多糖,主要被用于分子量的测定,但是通过研究我们发现这两种多糖在水溶液中的构象为一个球状线团,他们的MH系数α为0.5左右,而上面我们提到的香菇多糖,羧甲基纤维素,透明质酸,半乳甘露聚糖等高分子物质在水溶液中的结构为扩展性构象,因此当我们用Pullulan和Dextran这两种具有线团构象的标准品来测定这些具有扩展性链结构的大分子物质分子量时,是不是可以准确测定它们的分子量呢?为了进行对比研究,我们选择了另一种分子量测定标准品燕麦β葡聚糖(PS-OBG,来源于百特纯大分子有限公司),该标准品是从燕麦细胞壁中提取的,结构单元为葡萄糖,与Pullulan和Dextran相同,但它是由β-1,4和β-1,3键连接而成的线性结构,在水溶液中呈现扩展性构象,MH系数α为0.71.实验材料:待测样品:香菇多糖(PLE),羧甲基纤维素(CMC),半乳甘露聚糖(GG)葡聚糖标准品套盒:Shodex P-82,百特纯通用分子量套盒(PS-OBG-GMw)[img]http://ng1.17img.cn/bbsfiles/images/2008/12/200812160948_124375_1672347_3.jpg[/img]实验方法:GPC法和光散射(LLS)法实验结果:图一:P-82和PS-OBG-Mw的GPC图谱,水溶液中构象,MH系数对比图[img]http://ng1.17img.cn/bbsfiles/images/2008/12/200812160949_124377_1672347_3.jpg[/img]表一:三种不同物质分子量的测定结果SamplesMw1Mw2Mw3PLE707k1160k790kGG122k331k112kCMC130k354k148kMw1 : 用PS-OBG-GMw 套盒测定值Mw2: 用Pullulan-82 套盒测定值Mw3 : 用光散射测定值从表一中我们可以发现用P-82来测定这些具有扩展性链结构的大分子物质时,会导致明显的过高测定,之所以会出现这样的情况,是因为对于同样分子量的标准品来说,球形线团结构在GPC柱子中流出时间要比扩展性结构的流出时间长,而用PS-OBG-Mw来测定的话,测定值则非常接近光散射测定值,因此在对于大多数植物及真菌类多糖和具有扩展性链结构的大分子物质来说,用PS-OBG来测定是非常适合的。
[color=#444444]我想用液相分开两个大分子的聚合物,分子量都在40000左右,不同之处是其中一个加了四个小分子,试了十来根普通的柱子,没有效果,请各位大侠指教分离聚合物的色谱柱有哪些?很急, 谢谢![/color]
以前做的都是小分析化药,想问一下,在[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]方法建立和大分子样品生物样品提取时,和普通化药有啥区别(用的是岛津-AB4500/5500)大分子分子量大小有要求吗,是不是多大的分子都可以检测。我们的质谱质量轴超过2000Da时容易有偏差,请大神赐教、、、
[align=center][b][color=#33cc00]以叶黄素为例谈开发长链小分子化合物[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]方法[/color][/b][/align] [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181912494363_678_3425481_3.png[/img] [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181912490489_1922_3425481_3.jpg[/img] 小分子 蛋白/多肽 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181912497062_938_3425481_3.png[/img] [align=center] 长链小分子化合物[/align] 1.小分子本文描述小分子一般指分子量低于1000的化合物,为啥不讨论大分子化合物?因为APCI不太适合做蛋白/多肽等大分子。 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181912498331_515_3425481_3.png[/img] [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181912494991_1509_3425481_3.png[/img] ESI离子源(thermo) APCI离子源(rhermo) 2.常用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]离子源有ESI和APCI两大类型,做小分子方法一般情况都是ESI使用频率比较高,至于具体俩种类型离子源差异有兴趣可以百度一下(实在不想凑字数)。 常用四级杆有三重四级杆[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]、单四级杆[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url] 开发长链小分子化合物[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]方法常遇到以下两个难点: A.ESI源下注定响应不高 B.峰型不太理想 ESI源下响应低的情况几乎是肉眼可预见,长链小分子化合物大多带电位点少,就导致该类型化合物电离效率低,所以ESI源下响应就不会太高,而且某些特殊的长链小分子化合物ESI 源下还存在响应不稳定的情况(例如大部分类胡萝卜素)。针对此类情况其实更换APCI源就能很好的解决,此类化合物一般在APCI源下有稳定且较高的响应,在较高的响应下解决其他问题会比较方便,此策略可应用于多数带电位点少化合物。 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181912496642_6751_3425481_3.png[/img] 乙腈水下长链小分子化合物很难有正常峰型 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409181912497716_7598_3425481_3.png[/img] 使用异丙醇/甲醇-乙腈水流动相下化合物得到较好峰型 长链小分子化合物的结构直接导致此类化合物峰型不理想。链长,带氧官能团少,按常规的乙腈水去跑很可能就是一个鼓包,没有峰型,在流动相中添加适当比例的异丙醇,就能得到该化合物较好的峰型,对化合物的定性定量都有帮助。 以叶黄素为例谈开发长链小分子化合物[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]方法 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409182004172464_9821_3425481_3.png[/img] [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409182004173804_321_3425481_3.jpg[/img] 小分子 蛋白/多肽 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409182004170563_8516_3425481_3.png[/img] [align=center] 长链小分子化合物[/align] 1.小分子本文描述小分子一般指分子量低于1000的化合物,为啥不讨论大分子化合物?因为APCI不太适合做蛋白/多肽等大分子。 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409182004177510_1403_3425481_3.png[/img] [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409182004174416_7563_3425481_3.png[/img] ESI离子源(thermo) APCI离子源(rhermo) 2.常用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]离子源有ESI和APCI两大类型,做小分子方法一般情况都是ESI使用频率比较高,至于具体俩种类型离子源差异有兴趣可以百度一下(实在不想凑字数)。 常用四级杆有三重四级杆[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]、单四级杆[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url] 开发长链小分子化合物[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]方法常遇到以下两个难点: A.ESI源下注定响应不高 B.峰型不太理想 ESI源下响应低的情况几乎是肉眼可预见,长链小分子化合物大多带电位点少,就导致该类型化合物电离效率低,所以ESI源下响应就不会太高,而且某些特殊的长链小分子化合物ESI 源下还存在响应不稳定的情况(例如大部分类胡萝卜素)。针对此类情况其实更换APCI源就能很好的解决,此类化合物一般在APCI源下有稳定且较高的响应,在较高的响应下解决其他问题会比较方便,此策略可应用于多数带电位点少化合物。 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409182004176908_6402_3425481_3.png[/img] 乙腈水下长链小分子化合物很难有正常峰型 [img]https://ng1.17img.cn/bbsfiles/images/2024/09/202409182004183066_4822_3425481_3.png[/img] 使用异丙醇/甲醇-乙腈水流动相下化合物得到较好峰型 长链小分子化合物的结构直接导致此类化合物峰型不理想。链长,带氧官能团少,按常规的乙腈水去跑很可能就是一个鼓包,没有峰型,在流动相中添加适当比例的异丙醇,就能得到该化合物较好的峰型,对化合物的定性定量都有帮助。 叶黄素就是定型的长链小分子化合物,在使用乙腈水+ESI源时虽然也能找到母离子和对应子离子,但是峰型巨丑,稳定性也不理想,方法完全不能进行正常测样。更换APCI并在流动相中添加异丙醇后其峰型得到明显改善,方法得到极大改进,经验证此方法稳定性正常,能满足正常检测活动使用。
向大家请教个问题,做生物大分子如菌种代谢物(分子量约2万到3万)的核磁共振结构解析,是不是需要至少500MHz的磁场,做的过程中及解谱与化学上的核磁有些什么差异?多谢高手指点。
生物大分子分离【分享】[~144928~]
刚做了个酰胺类的大分子量化合物(分子量将近1000),过程中遇到很多问题,拿出来和大家探讨下,以求共同进步! 1)在正常的酸性(ph 4-7)流动性体系中,化合物峰拖尾严重,排除原因 既不是过载,又不是柱子坍塌(坏掉)或死体积引起的,更不是溶解样品的溶剂引起的,那么最终原因是什么?是因为酸性吸附?还是分子量大,在固定相和流动性中交换缓慢,进入固定相后很难出来? 2)在正常的碱性(ph9-11)流动性体系中,化合物峰型很完美,可惜残留很大,进一针样品后,需要再走N针空白试剂才能把柱子上的样品完全洗脱下来,而且在洗脱过程中,基线非常高。和酸性流动性相比,保留时间偏后!那么为什么残留如此大?是因为C18柱上的硅醇键对化合物有吸附作用?还是化合物在分子状态下,极性很小,正常的ACN流动性无法洗脱?
经常见到资料说,反相色谱法中,亲水性大分子聚合物(蛋白质、核酸等)难以从色谱柱中洗脱,甚至是永久性的。反相流动相的极性较大,且多数水组分含量较高,按理应该更容易洗脱吧请问原因何在?
【序号】:2【作者】:王凯【题名】:生物大分子魔芋的阳离子化改性及聚电解质复合物的研究【期刊】:武汉理工大学 【年、卷、期、起止页码】:2018【全文链接】:[url]https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CMFD&dbname=CMFD201902&filename=1019832452.nh&uniplatform=NZKPT&v=i8NcTeI2Rq255i6qGIcKWA1kDyDnxA8Dj7sYIKgA_k8UMTx7lV5MqdIHqkaNG2WA[/url]

[align=center][b]电喷雾质谱(ESI-HRMS)在生物大分子检测中的应用[/b][/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]GLP 耿建瑞[/align][align=center] [/align]电喷雾离子源(ESI)的工作原理如图1.7所示,雾化器的喷针加有高电压,液态分析物质低速通过喷针时,在高压电场的作用下,形成泰勒锥,当泰勒锥表面张力小于静电斥力时,形成雾状液滴,含同种电荷的的液滴在高压电场的作用下向毛细管移动,高温氮气干燥气反向吹动,使液滴不断挥发,直至产生脱溶剂,分析物以气态离子进入毛细管。[align=center][img=,660,288]http://ng1.17img.cn/bbsfiles/images/2017/09/201709081539_01_2904018_3.png[/img] [/align][align=center]图1. 电喷雾离子源(ESI)原理示意图[/align]ESI是一种“软电离技术”,在样品离子化的过程中,没有热汽化的过程,没有离子碎片的产生,能够保持样品丰度完整性;可以准确测出分子质量(mw),从而直观的区分出混合物中的不同组分;另外,ESI可以产生多电荷峰,而质谱测定的是质荷比,大大拓宽了仪器可检测的分子量范围,非常适合大分子化合物以及非共价复合物的分析。ESI也可用于核酸以及核酸与配体的相互作用的分析,与蛋白质的检测不同的是核酸的检测一般采用负离子模式。因为蛋白质含有较多的含氮碱基,在酸性及中性溶液中很容易质子化,而核酸含有磷酸基团,在溶液中容易失去质子,所以核酸的检测一般采用负离子模式。ESI离子源可以产生多电荷离子峰,使得质荷比大大降低,从而扩展了质谱的检测范围,可以检测蛋白质、核酸等大分子。但是同一化合物会产生多种电荷的质谱峰,不同的离子峰必须归属到能够产生该多电荷离子峰的离子,这是一个很复杂的过程。对于低分辨率的质谱,当质谱产生的多电荷质谱峰电荷较少时,每个质谱峰所带的电荷数可通同一离子的带不同电荷的离子峰的比值来确定。但是对于更复杂的样品如蛋白质、DNA等,由于其所含成分多,碎片离子也多多,生成的多电荷质谱情况很复杂——峰数目多,且会有峰折叠,掩盖等情况,其电荷数数和质量数的的确定就很困难。对于复杂样品的检测,提高质谱的分辨率则非常重要,对于一簇多电荷离子峰,我们可以通过相邻两个同位素峰的间距很容易的得到该离子峰所带电荷数,从而很计算出生成该多电荷峰的离子的分子量。但是生物样品成分复杂,即使一次小小的实验也可能会产生成千上万的质谱数据。这就需要对这些多点和质谱数据进行解卷积,对于质谱来说,“解卷积(Deconvolution)”是指利用电荷只能是整数的特点,将同一分子量的不同质荷比的峰按一定的算法组合到一起。科学家们开发出各种各样的算法及软件用于质谱数据的自动解卷积。但是各个仪器公司的解卷积软件价格昂贵,很多实验室,虽然可以利用ESI-HRMS可以得到大量的质谱数据,但是苦于无法解卷积,导致实验数据无法进行分析。在此,介绍由Horn等人提出一种基于高分辨数据的解卷积算法THRASH[sup][[/sup][url=#_ENREF_65][sup]65[/sup][/url][sup]][/sup]。THRASH的基本原理如图1.8:1,使用者设定质荷比m/z范围,质谱峰可能带电荷数目,信噪比阈值等信息。2,程序自动将高于指定信噪比阈值的质谱峰挑出。3,按照自相关算法[sup][[/sup][url=#_ENREF_66][sup]66[/sup][/url][sup]][/sup]计算出质谱峰的电荷数,并计算出平均分子质量。用第2步得到的平均分子质量结合Mercury算法 得出质谱峰簇的理论轮廓图[sup][[/sup][url=#_ENREF_67][sup]67[/sup][/url][sup]][/sup],4,将理论轮廓图与质谱中实际轮廓图对比,得到一个匹配分数(fit score),5,将高于fit score的质谱峰删掉,低于fit score的质谱峰重复第3步骤。[align=center][img=,655,494]http://ng1.17img.cn/bbsfiles/images/2017/09/201709081539_02_2904018_3.png[/img] [/align][align=center]图2高分辨质谱数据解卷积原理[/align]Navdeep等人基于THRASH开发的开源软件Decon2LS,对质谱数据进行解卷积可以生成电荷数(charge)、单一同位素峰(monoisotopic mass)、同位素最高丰度离子峰(most abundant isotope intensity)、平均分子量(average mass)以及对应的质谱丰度(abundance)等信息,为ESI-HRMS研究蛋白质、DNA及其与小分子化合物的相互作用的分析提供了详细的数据。
请问这类核磁一般要扫描测试多长时间呢?听说通常解出一个生物大分子的结构所需要时间是2个多月?这部分的时间大部分都花在解谱上么?
对于含CHO的大分子物质,有时候结构复杂,比如含很多环状结构,那么这时候怎么知道它的极性大小呢,有没有测评物质极性的软件呢?请专家帮下忙,谢谢!
RT.也是今天突然想到的。从我自己的操作来看,大分子蛋白质分子在反相柱中似乎是一种全或无的保留行为(动或者不动)。也就是假设X蛋白在40%乙腈:60%水的时候保留时间是7分钟,如果我用35%乙腈的可能冲1个小时它都纹丝不动,反之,我一上来就用60%的冲,保留时间还是7分钟,没有明显的加速。所以想在这里请教一下大分子蛋白质在反相柱中的保留行为和分配理论,常规的理论塔板似乎不太适用。
“生物大分子多维核磁共振”一书由夏佑林,吴季辉,刘琴及施蕴渝编著,中国科学技术大学出版社出版。该书介绍了多维核磁共振波谱学基本原理及其在结构生物学中的应用。全书分为13章,内容包括核磁共振基本理论,一维多脉冲实验,二维NMR基本原理,蛋白质结构测定,蛋白质的稳定同位素标记,三维四维NMR波谱,蛋白质折叠,酶反映机理研究,核酸和糖的结构测定,各种选择性实验,膜和膜蛋白的固态NMR研究以及核磁共振成像。该书参考了国内外一些核磁共振优秀教材的内容,并作了很好的归纳总结。
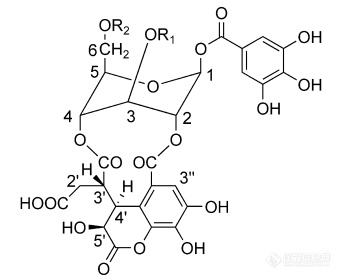
化合物特征:大分子、不易吸附于C18填料。极性较大,在醇溶液中溶解良好,水中也可溶。酚羟基含量多,易与金属离子发生反应。样品溶液粘度较大,干燥后手碾有类似于糖类物质的黏滞感[img=,342,279]https://ng1.17img.cn/bbsfiles/images/2019/10/201910091234058072_7433_1773263_3.png!w342x279.jpg[/img]
安捷伦gcms大分子多环芳烃,比如晕苯怎么才能测到。升温程序,scan方法有什么建议吗?
测试目的:通过样品前处理将样品中的大分子量杂质富集,然后做GCMS进行外标法定量现在不知道如何富集样品中的大分子量杂质,我的样品极性都很弱,样品结构是饱和环己烷接烷基 或者饱和环己烷接苯环类的;样品的分子量200~500;样品中有痕量大分子量杂质,分子量500~2000,大分子量杂质可能是样品的寡聚物 或者是塑料制品的寡聚物。现在需要寻找样品前处理方法,将样品中痕量大分子量杂质富集起来。目前已知行业内是将25克样品进行前处理,前处理后做GCMS进行定量,但是不知道前处理方式。看到药典中有用凝胶色谱富集药品中寡聚物或高分子,但是我的分子量相差这么低,大分子量杂质的极性也是很弱的。请大神们多多指教。
为了促进国际学术交流,研讨生物大分子核磁共振研究以及技术的最新进展,将于2009年6月25日到6月29日在中国科学技术大学生命科学学院召开“生物大分子核磁共振研讨会,2009”,会议由中国科学技术大学承办。会议将交流生物大分子核磁共振领域最新的研究方法和国际研究前沿及发展动态。报告内容包括, 应用液体,固体核磁共振方法研究生物大分子(包括水溶性蛋白及其复合物,核酸,膜蛋白)的三维结构和不同时间尺度下的动力学分析,应用核磁共振方法进行先导药物筛选,候选药物优化,及代谢组学分析等相关领域的理论,方法,技术及其在生物化学,细胞生物学,药理学等生物学领域应用的未公开发表过的研究工作。会议组委会已经成功邀请到美国国家科学院院士,美国国立卫生研究院研究员,著名核磁共振科学家Ad Bax博士作会议主题报告。近30位国际,国内知名华人核磁共振科学家(包括学术界,工业界)已经应邀出席研讨会并作学术报告。具体研讨会信息请见网站:http://bionmr.ustc.edu.cn/snbm2009 附件是第一轮通知,我们诚挚地欢迎您参加此次研讨会。并请将本通知传达给感兴趣的同行,同学!非常感谢!田长麟研讨会秘书长02/16/2009
怎样纯化大分子蛋白 26kd不需要量很大。可以打质谱就行。
[b][font='微软雅黑',sans-serif][color=black][back=white]【序号】:3【作者】: 何扬芳【题名】:应用质谱技术提升传统中药的质量控制和生物大分子药物的结构表征能力[/back][/color][/font][/b][align=left][font='微软雅黑',sans-serif][color=black][back=white]【期刊】:吉林大学 博士论文[/back][/color][/font][font='微软雅黑',sans-serif][color=black][/color][/font][font='微软雅黑',sans-serif][color=black][back=white]【年、卷、期、起止页码】:2020[/back][/color][/font][font='微软雅黑',sans-serif][color=black][/color][/font][font='微软雅黑',sans-serif][color=black][back=white]【全文链接】:[/back][/color][/font][url=https://kns.cnki.net/kcms2/article/abstract?v=3uoqIhG8C447WN1SO36whLpCgh0R0Z-iVBgRpfJBcb4JAybTo8M4ljH5Ce6ATfKqWkZZyuKToFWj_RADn7Nr0YNPrffgey8c&uniplatform=NZKPT]应用质谱技术提升传统中药的质量控制和生物大分子药物的结构表征能力 - 中国知网 (cnki.net)[/url][/align][align=left] [/align]
测试目的:通过样品前处理将样品中的大分子量杂质富集,然后做GCMS进行外标法定量现在不知道如何富集样品中的大分子量杂质,我的样品极性都很弱,样品结构是饱和环己烷接烷基 或者饱和环己烷接苯环类的;样品的分子量200~500;样品中有痕量大分子量杂质,分子量500~2000,大分子量杂质可能是样品的寡聚物 或者是塑料制品的寡聚物。现在需要寻找样品前处理方法,将样品中痕量大分子量杂质富集起来。目前已知行业内是将25克样品进行前处理,前处理后做GCMS进行定量,但是不知道前处理方式。看到药典中有用凝胶色谱富集药品中寡聚物或高分子,但是我的分子量相差这么低,大分子量杂质的极性也是很弱的。请大神们多多指教。以前注册使用账号忘记了,新注册的账号,积分很少,还请大神笑纳。
生物大分子溶液的粘度测定怎么测,用那种粘度计?
本人对X射线晶体学很陌生。请问:1. 当前X射线仪器主要又哪几种。2. 测定生物大分子(蛋白质)主要应用哪种仪器,是X射线衍射仪吗?
首先,对于复杂化合物,如分子量特别大的多糖,在样品制备时要注意样品的溶解度,有时分子量过大的多糖溶解度不好,在H谱和C谱中有些样品的特征信号峰未呈现出来(分子量大的有的弛豫也很快,在一维二维中的信号有差异),再加上C谱的灵敏度要比H谱低,所以这类物质在样品制备时要提高样品浓度,可以考虑超声助溶等方法,效果特别不好的可以考虑水解后再检测; 其次,在信号归属中,大分子量的化合物很多的峰会出现重叠现象,堆积在3.0~4.0之间(H)、60~80(C),峰重叠的现象很严重,这时我们要提高样品的纯度,尽量降低其他杂质化合物对样品测试的干扰,同时考虑提高样品的浓度; 结合二维核磁,我们可以分析出多糖的单糖残基顺序、单糖残基在糖苷键中的位置、环状结构的类型和糖苷键的构型等信息,具体分析一定要结合样品相关文献进行,通常情况如下: 1、COSY:可以从容易辨认的质子(异头质子、甲基质子)开始,寻找糖上其他碳位的质子,归属每个质子信号; 2、HSQC:可找出易头碳质子相连的C信号及糖基上每个质子连接的C信号,通常可结合甲基化分析和其他二维谱; 3、HMBC:可联系2~4个共价键的H核和C核,可以以列表的形式写下关联的位点,从结构简单或有特色的峰开始,一一对应,实际处理时,并不是每个H都可以找到旁边所对应的C,所以要结合样品相关的文献进行分析。







 我要推广仪器
我要推广仪器
 下载APP
下载APP