氨唑草酮(BAY314666)是拜耳公司1988年发现的三唑啉酮类除草剂,1999年在英国布莱顿世界植保大会上推出。氨唑草酮为光合作用抑制剂,敏感植物的典型症状为褪绿、停止生长、组织枯黄直至最终死亡,与其它光合作用的抑制剂(如三嗪类除草剂)有交互抗性,主要通过根系和叶面吸收。具有内吸活性,通过抑制敏感植物的光合作用,干扰正常的电子传递。通常使用三到四周就能产生效果。http://ng1.17img.cn/bbsfiles/images/2017/04/201704222147_01_1623180_3.jpeg氨唑草酮欧洲专利EP0370293,已于2009年11月3日到期;美国专利US5194085,已于2010年5月15日到期。氨唑草酮的适用对象主要为甘蔗、玉米和草坪。它可以有效防治玉米和甘蔗上的主要一年生阔叶杂草和禾本科杂草。在玉米上,氨唑草酮对苘麻、藜、野苋、宾州苍耳和甘薯属等具有优秀防效,此外对甘蔗上的泽漆、甘薯属、车前臂形草和刺蒺藜草等也有很好的防效。甘蔗玉米在我国种植面积较大,因此氨唑草酮在我国的应用市场也比较广阔。氨唑草酮的最大优点是有抗旱和非毒性特性,应用更灵活,能降低工作量,减少整修,降低杀菌剂、杀虫剂和植物生长调节剂的使用。除草时间长,对地下水安全,对后茬作物安全,用量仅为莠去津的1/2—1/3,也因此成为了高毒农药莠去津等的最佳替代产品。与莠去津相比,氨唑草酮原药成本较高。另据业内人士透露,该产品在玉米作物中使用尚存安全性问题,因此目前国内市场上并无氨唑草酮产品的销售。
药典规定甘草酸单氨盐灰分精品不超1/1000,但我用做瓷坩埚做出来老是负数,估计是坩埚材质不行,请问要用什么材质的坩埚来做?附检测条件:800度,4小时。刚毕业从事检测工作,请各位帮帮。小弟先谢过。
请问有人做过氨唑草酮的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]方法,
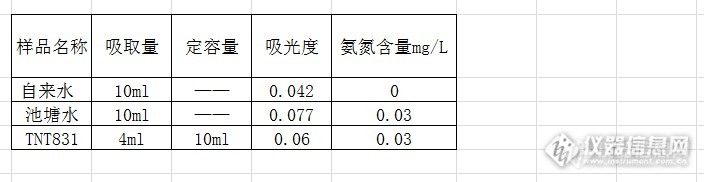
老久没有动手了,老久没有玩紫外了。老胳膊老腿还利索否?本次qingqingcao向分光进军。玩会儿分光光度计。某公司是生产水质监测仪的,最近在搞氨氮免费试剂的测试。恰好我们学校也在弄这方面的检测,随机填写了表格,请某公司给我们寄点免费质控样。看看我的水平如何?http://ng1.17img.cn/bbsfiles/images/2013/09/201309240912_466715_1626663_3.jpg啥都自己来吧。反正配制试剂,标准品都是小菜一碟。看我的。首先介绍下氨氮测试的意义和方法。氨氮是指水中以游离氨(NH3)和铵离子(NH4+)形式存在的氮。动物性有机物的含氮量一般较植物性有机物为高。同时,人畜粪便中含氮有机物很不稳定,容易分解成氨。因此,水中氨氮含量增高时指以氨或铵离子形式存在的化合氨。根据国标,氨氮测试使用三种方法。1)纳氏试剂分光光度法。2)酚盐分光光度法。3)水杨酸分光光度法。我们使用的是第三法。这个方法主要还是用比色方法来测试的。其原理简单来说,是由于亚硝基铁氰化钠存在下,氨氮在碱性溶液中与水杨酸盐—次氯酸盐生成蓝色化合物,色度与含氮量成正比。首先就是试剂的配制啦。这里省略数字。没啥好写的。http://ng1.17img.cn/bbsfiles/images/2013/09/201309240913_466716_1626663_3.jpg其次,标准品的配制。这里好好写写。称取105℃烘制1小时的氯化铵。0.4211g到100ml容量瓶中。超纯水定容摇匀。吸取该溶液0.5ml到100ml容量瓶中,超纯水定容摇匀。此时工作液氨氮浓度为5.513mg/L http://ng1.17img.cn/bbsfiles/images/2013/09/201309240920_466722_1626663_3.jpg接着准备8根10ml比色管。列队放好。分别加入工作液0
草甘膦、氨甲基膦酸和草甘膦内标衍生: 取1ml 20g/L上述物质标准溶液(水作溶剂),分别加0.5ml 50g/L硼酸钠、0.5ml 10g/LFMOC-Cl衍生剂,37℃衍生2h。SN/T 1923是缓冲盐和衍生剂各加0.2ml,并且衍生剂浓度是1g/L,担心衍生不完全我多加了,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS上扫描啥也没有或者说响应极低不稳定,倒是有几个离子响应很稳,比如草甘膦扫出来是179,365和397,正常来说179、392。 后面,我又直接拿草甘膦和氨甲基膦酸(未经衍生)上机扫描,文献上有负离子扫描出峰的,我试了一下,草甘膦(分子量169)扫出来是168,响应较低不稳定,但合理。就是说仪器没问题,标准品没问题。 问题来了,到底是哪里出了问题?衍生剂我都是临配现用的,顶多也是过量了而已
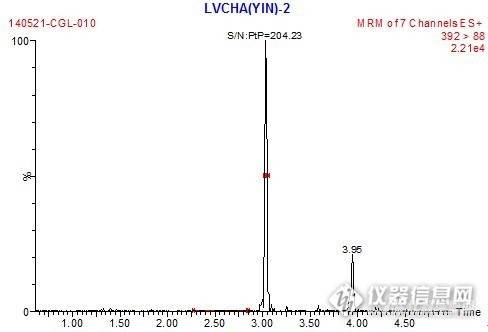
【生活中的仪器分析】样 品:蔬菜、水果、茶叶、茶粉等食品检测项目:草甘膦、草铵膦、氨甲基膦酸参考标准:SN/T 1923-2007检测仪器:a.WATERS液相色谱串联质谱仪:配有电喷雾(ESI)离子源(可用其他品牌作用等效的高效液相色谱质谱仪替代)b.Biotagevacmaster固相萃取仪c.IPRE Qclean PMG草甘膦专用固相萃取柱d.BiotageTurbovap LV 快速浓缩仪e.IKA MS3 涡旋混匀器g.TOMY-MX307离心机g.昆山超声波清洗器实验过程:1.提取及预处理称取2-5g(精确到0.001g)试样于50 mL聚丙烯离心管中,加入100μL内标液,加入20.0 mL水超声提取30min,于10000 r/min离心5min,取1.0 mL上清液于2mL子弹头离心管中,加入100μL酸度调节剂(注A),涡旋混匀,15000r/min离心5 min,待净化。注A:酸度调节剂配制方法:纯水+色谱纯甲醇+盐酸=160+40+13.4(V/V/V)2.固相萃取净化І将PMG-І柱(蓝柱)用2mL甲醇和2 mL 0.5%甲酸淋洗活化并自然滴干,将加入酸度调节剂处理的提取液(2)转移到小柱上,用5mL刻度试管收集流出液(1-2滴/秒),用1.0mL 0.5%甲酸洗柱并真空抽干,合并流出液,用移液枪吸取50%NaOH调pH7-9(用1-14pH试纸,根据样品不同约20-50μL),加水定容到3 mL刻度,混匀,待衍生。3.衍生步骤准确吸取600 μL净化液(3)于2mL子弹头离心管中,加入200 μL 5%硼砂溶液,边涡旋,边加入200 μL 25g/L FMOC-Cl乙腈溶液(注B),放置10min,加入50 μL甲酸,涡旋混匀,15000 r/min离心5min,吸取上清液准备过PMG-ІІ柱。注B:25 g/L FMOC-Cl乙腈溶液配制方法:称取0.25 gFMOC-Cl,溶解于10 mL色谱纯乙腈中。4.固相萃取净化ІІ 将PMG-ІІ柱(红柱)用2 mL甲醇和2 mL 0.5%甲酸淋洗并自然滴干,将上清液过PMG-ІІ柱,用3 mL水淋洗小柱,真空抽干5-10 min,再加入2 mL正己烷淋洗小柱,滴干后真空抽干5 min,最后用5 mL 5%氨水/甲醇洗脱小柱(1-2 mL/min)并用5 mL刻度试管收集流出液,45℃,氮气吹至近干,用20%乙腈定容1.0 mL,涡旋混匀,过0.2 μm PTFE膜后上机测试。5.测定5.1色谱条件a.色谱柱:Waters BEH-C18,1.7 μm,2.1 mm×100 mm;b.流动相:5mmol/L乙酸铵:乙腈梯度洗脱,梯度表见表1; 表1 流动相及梯度 时间(min)流速(mL/min)5mmol/L乙酸铵(%)乙腈(%)00.3901020.362384.40.362384.50.35956.50.35956.60.390109.00.39010c.检测器:串联四极杆质谱仪;d.柱温:35℃;e.进样量:10 μL。5.2质谱分析条件a)电离源:电喷雾正离子模式;b)毛细管电压:3.50KV;c)源温度:120℃;d)脱溶剂气温度:400℃;e)脱溶剂气流量:700L/h;f)碰撞室压力:2.7í10-3mbar;g)特征离子及参数见表2。 表 2 草甘膦和氨甲基膦酸的主要特征离子 化合物保留时间(min)母离子+(m/z)锥孔电压(V)子离子(m/z)碰撞能量(eV)草甘膦1.32392.215*88.02515214.01
请问 哪位老师做过草甘膦和氨甲基膦酸[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]MS检测,求前处理和仪器检测方法?
中国农药信息网: 百草枯排放标准限值的确定及意义百草枯生产企业目前执行的排放标准为1996年颁布的《污水综合排放标准》(GB8978-1996)以及某些地方自定的排放标准。这些现行排放标准没有考虑到百草枯行业的特殊性,标准指标和限值的设定没有针对性。指标不足以反应废弃物的危害,对危害严重的物质未加控制。新标准对特征污染物吡啶类和百草枯规定了严格的排放限值,将促进企业采用排污少、清洁的先进生产工艺,从而减少污染物的排放。 百草枯是一种触杀型灭生性联吡啶类除草剂。1958年由ICI公司首先开发成功,1961年ICI公司首次实现工业生产。百草枯是近年来在国内发展较为迅速的农药品种之一,由于其作为除草剂具有药效高、用量低、起效快等特点,深受广大农户的欢迎。 百草枯生产过程中产生含有吡啶类物质及百草枯阳离子。吡啶类属于难降解有毒化合物,百草枯是具有药效的活性物质。因此,本标准选择上述成分作为特征污染物,并规定了新源及现源的排放浓度限值。 根据目前国内企业治理水平及百草枯对环境的影响,标准值中现源特征污染物吡啶为5毫克/升,百草枯离子为0.1毫克/升;新源依据国内外最佳治理技术,吡啶排放标准为2毫克/升,百草枯离子为0.03毫克/升,现有企业可在一定时间内提升治理技术水平,达到新源标准。而在特别地区,现有企业和新建企业的排放限值均为吡啶1毫克/升,百草枯离子0.01毫克/升。 本标准根据百草枯生产工艺的特点及目前国内外治理技术,规定了单位基准排水量,达到了总量控制的目的,使标准具有更强的可操作性。 由于百草枯生产过程中产生高浓度、高毒性废水,难于处理,许多中小企业由于资金、技术等方面的原因,甚至没有采取任何环保措施。目前采用焚烧工艺是解决水污染的最佳技术。新标准的实施,将促进焚烧工艺治理百草枯废水技术的推广。 本标准在编制过程中也体现了淘汰落后工艺、促进清洁生产的原则。百草枯生产工艺有高/中温钠法、低温钠法、水氰法、醇氰法工艺和氨氰法。高/中温钠法使用金属钠,反应过程易发生燃烧和爆炸事故、产品收率通常仅为45%~60%,很容易生成强致癌性的三联吡啶类物质,污水产生量大。低温钠法则具有系统平稳、三联吡啶等杂质的产生量少、总收率为90%~95%、三废量少或基本无污水产生等许多优点。但该工艺对冷冻等设备要求高,控制条件苛刻,能耗和成本较高,国内还没有企业采用这种工艺。目前国内普遍采用的是氨氰法和醇氰法。氨氰法工艺由于操作简单、平稳,没有起火和爆炸的危险,同时工艺中所有氨溶液都可循环使用,产品质量稳定,不产生有害物质,收率可达95%以上,被认为是先进的工艺。本标准中规定了三联吡啶不得检出,因而限制了高/中温钠法工艺生产。
从己内酰氨与二甲苯的混合液中分离出己内酰氨?
草甘膦和氨甲基膦酸与三氟乙酸酐和七氟丁醇衍生反应,到底生成的衍生物分别是什么?
求助:茶叶中除草剂吡氟酰草胺(DlFLUFENICAN)的检测方法。
各位大侠们,请问谁有草酰氯的检测方法啊,给我发一份可以吗我的油箱sheny123@tom.com 谢谢啊!
我用安捷伦的1290-6460检测茶叶中的草甘膦、氨甲基膦酸,标准是SN/T 1923,可是母离子、子离子都找不到啊,麻烦大家指点一下!
磺酰脲类除草剂的开发始于上世纪70 年代末期。70 年代初, 美国杜邦公司的G. Levitt博士发现磺酰脲类化合物4- 氰基苯基苯磺酰脲, 在2kg a.i./hm2 剂量下有弱的植物生长阻滞作用, 于是将其作为先导化合物进行结构优化, 合成了一系列该类化合物。发现由芳香基、磺酰脲桥和杂环3 部分组成, 其基本化学结构式在每一组分上取代基的微小变化都会导致生物活性和选择性的极大变化。杜邦公司在Levitt 博士的指导下, 经过不懈努力, 终于在1978 年研制出第1 个磺酰脲类除草剂氯磺隆,并于1982 年商品化。氯磺隆以极低用量进行芽前土壤处理或苗后茎叶处理, 可有效地防治麦类与亚麻田大多数杂草。 氯磺隆问世之后, 除杜邦公司外, 瑞士汽巴- 嘉基、日本的石原产业、日产化学、武田、德国拜耳、美国氰胺等农药公司和韩国化学研究所、我国南开大学元素有机化学研究所等也进行了该类除草剂的研制和开发。甲磺隆、甲嘧磺隆、氯嘧磺隆、苯磺隆、噻吩磺隆、苄嘧磺隆等一系列产品随后相继问世, 目前, 大约已有30 个实现商品化。特点:1 超高效、广谱、高选择性、低毒的优良品质催生市场快速发展,此类除草剂超高效, 用量以g/hm2 计, 其生物活性超过传统除草剂100~1 000 倍, 使除草剂的发展进入“超高效时代”。此类除草剂再一个优势是广谱、高选择性, 对许多一年生或多年生阔叶、禾本科杂草和莎草, 尤其是阔叶杂草有特效, 已广泛用于水稻、麦类、大豆、玉米、油菜等多种作物、草坪和其他非耕地。此外, 它们对哺乳动物和鱼类毒性较低,Ames 试验阴性, 不致畸、致癌、致突变, 在环境中易分解, 这意味着是一类环境友好的除草剂。所以, 它们问世之后就发展极快, 有些已成为一些作物田的当家除草剂品种。而且,新的品种还在不断地商品化。1996 年这类除草剂的销售额就达到了15.05 亿美元, 仅次于有机磷类除草剂, 其中苄嘧磺隆、烟嘧磺隆、氟嘧磺隆和噻吩磺隆4 种产品的销售额分别为2.6 亿、1.5 亿、1.5 亿和1.3 亿美元。随着存在环境问题除草剂的淡出市场, 磺酰脲类除草剂得到了快速的发展, 目前, 在世界农药市场中占有举足轻重的重要地位。近年来, 我国多种因素促成除草剂市场快速发展, 麦类、玉米、甜菜等旱田作物及稻田除草剂使用量大幅上升, 市场扩大, 给该类除草剂的发展提供了良好的发展机遇。2 世界主要磺酰脲类除草剂产品磺酰脲类除草剂在我国使用广泛、使用时间长, 推广比较成功的有杜邦的苯磺隆、苄嘧磺隆、烟嘧磺隆、玉嘧磺隆、噻吩磺隆等, 它们在我国的推广使用超过10 年。销售额较高的, 如杜邦的苯磺隆和苄嘧磺隆。石原的烟嘧磺隆在上个世纪90 年代初就在我国推广使用,在我国的麦类、玉米除草中去得了好的效果。
上世纪60年代。当时,杜邦公司开发出了首个脲嘧啶类除草剂—除草定,正式开启了该类除草剂研发的先河。而真正掀起脲嘧啶类除草剂开发热潮的是在上世纪90年代,当时人们对于该类除草剂的作用机理有了更深入的了解,发现脲嘧啶类除草剂属于原卟啉原氧化酶(PPO)抑制剂。杜邦公司在推出除草定后,又相继推出了异草定和特草定等产品。富美实的双苯嘧草酮以及先正达的氟丙嘧草酯均属于该类除草剂。而巴斯夫于2009年推出的苯嘧磺草胺(saflufenacil)更属于该类除草剂中的佼佼者。苯嘧磺草胺能够适用于多种生产系统和非耕地,在苗后或苗前均能使用;其次,适用作物多。苯嘧磺草胺能够用于包括谷物、玉米、棉花、水稻、高粱、大豆和果树等在内的30多种作物上;再次,防除谱广。苯嘧磺草胺能够防除90余种阔叶杂草,包括一些对三嗪类、草甘膦及乙酰乳酸合成酶抑制剂存在抗性的杂草。另外,它也具有作用快、残效期长等多种特性。http://ng1.17img.cn/bbsfiles/images/2017/02/201702010042_01_1623180_3.jpg2009年,苯嘧磺草胺在南美国家尼加拉瓜、智利和阿根廷三国登记。2010年,苯嘧磺草胺与精二甲吩草胺的复配制剂Verdict在美国获得登记,用于大豆。同年,苯嘧磺草胺正式登陆中国,以70%水分散粒剂(商品名:巴佰金)的形式面世,用于柑橘园和非耕地的杂草防除,由诺普信负责在中国市场的总经销。目前,苯嘧磺草胺已在美国、加拿大、中国、尼加拉瓜、智利、阿根廷、巴西和澳大利亚等国登记。苯嘧磺草胺可替代苯氧类除草剂2,4-D和磺酰脲类除草剂与草甘膦复配,可降低防治顽固性杂草对草甘膦的使用量。2014年,苯嘧磺草胺的全球销售额达到1.4亿美元。据巴斯夫公司预测,苯嘧磺草胺可实现3亿欧元的年峰值销售额。苯嘧磺草胺目前仍处于专利保护期中,其在中国的专利为巴斯夫于2001年申请的《尿嘧啶取代的苯基氨磺酰羧酰胺》,专利号为ZL01801896.3,对苯嘧磺草胺的化合物及合成方法进行了保护,该专利将于2021年4月30日到期.
有草酰氯气相如何检测 需要柱前处理吗?如何处理?谢谢各位大侠!!

“超高效液相色谱-串联质谱法测定大豆中草甘膦及其代谢物氨甲基膦酸的残留”是本人去年开展大豆中草甘膦检测项目整个试验过程的总结,欢迎各位老师和同行批评指正,该文章还未在任何刊物上发表。[align=center][b]超高效液相色谱-串联质谱法测定大豆中草甘膦及其代谢物氨甲基膦酸的残留[/b][/align][align=center][/align][align=center]户江涛[/align][align=center](黑龙江省农垦科学院测试化验中心,黑龙江 佳木斯 154007 )[/align]摘要:采用超高效液相色谱-串联质谱法建立了快速检测大豆中草甘膦和氨甲基膦酸残留量的分析方法。试样经水超声提取,二氯甲烷去除脂肪,C[sub]18[/sub]固相萃取柱净化后,在硼酸钠缓冲溶液中与9-芴甲氧羰酰氯(FMOC-Cl)进行衍生反应,其衍生产物在C[sub]18[/sub]色谱柱上以 2 mmol/L 乙酸铵溶液和乙腈为流动相,进行液相色谱分离:质谱检测采用电喷雾正离子化模式和多反应监测模式(MRM)。结果表明,草甘膦和氨甲基膦酸在0.001~0.5 mg/L范围内线性关系良好,相关系数(R)分别为0.9996和0.9993,定量限(LOQ)均为0.01mg/kg。在空白大豆样品添加浓度为0.02、0.2、2 mg/kg 时,草甘膦和氨甲基膦酸的平均回收率分别为80.2%~91.5%和77.7%~89.3%,相对标准偏差(RSD)分别为3.37%~6.96%和4.11%~8.27%(n=6)。本方法快速、简便、灵敏,适用于大豆中草甘膦和氨甲基膦酸残留的同时检测。关键词:超高效液相色谱-串联质谱;大豆;草甘膦;氨甲基膦酸;衍生反应[align=center]Determination of glyphosate and its metabolite aminomethyl-phosphonic acid residues in soybean by ultra performance liquid chromatography-tandem mass spectrometry[/align][align=center]HU Jiangtao[/align][align=center]([i]Testing and Analysis Center of Heilongjiang Academy of Land Reclamation Sciences, Jiamusi 154007,China[/i])[/align][b]Abstract:[/b]A method[b] [/b]was developed for the determination of glyphosate(PMG) and aminomethyl-phosphonic acid(APMA) residues in soybean by ultra performance liquid chromatography-tandem mass spectrometry(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS). After extracted with water under ultrasonication, the sample was defatted with dichloromethane and purified by C[sub]18 [/sub]solid phase extraction cartridge, and then PMG and APMA were derivatized using 9-fluorenylmethoxycarbonyl(FMOC-Cl) in borate buffer for 2 h.The derivatives of PMG and APMA were separated on a Waters BEH C[sub]18[/sub] column with gradient elution with the mobile phase of 2 mmol/L ammonium acetate and acetonitrile, and finally detected by positive eletrospray ionization-mass spectrometry(ESI[sup]+[/sup]-MS/MS) in multiple reaction monitoring(MRM) mode.The results showed the linearities of PMG and APMA were good in the concentration range of 0.001~0.5 mg/L ,and the correlation coefficients were 0.9996 and 0.9993 respectively. The limit of quantification(LOQ) of PMG and APMA was both 0.01mg/kg. At the spiked levels of 0.02、0.2、2 mg/kg in the blank soybean samples, the mean recoveries of PMG and APMA were 80.2%~91.5% and 77.7%~89.3% respectively, and the relative standard deviation(RSD) of PMG and APMA were 3.37%~6.96% and 4.11%~8.27% res-pectively(n=6).This method is fast,simple,sensitive, and suitable for simultaneous determination of PMG and APMA in soybean.[b]Key words: [/b]ultra performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) soybean glyphosate(PMG) aminomethyl-phosphonic acid(APMA) derivatization草甘膦(Glyphosate,PMG)又名镇草宁、农达,分子式为C[sub]3[/sub]H[sub]8[/sub]NO[sub]5[/sub]P,是1971年美国孟山都公司研发的一种有机磷除草剂,因其兼具内吸、传导性、灭生性及非选择性,同时不易在生物体内累积,故广泛应用于农业生产中一年生和多年生杂草防除,是目前世界上应用最广、生产量最大的除草剂[sup][/sup]。草甘膦及其在植物中的主要代谢物氨甲基膦酸(Aminomethyl-phosphonic acid,APMA,分子式为CH[sub]6[/sub]NO[sub]3[/sub]P)均属于强极性、易溶于水的高沸点化合物,具有不易挥发、无紫外吸收等特性,因此用常规方法分析检测十分困难[sup][/sup]。 目前, PMG和APMA残留检测的方法主要有色谱法(GC[sup][/sup]、LC[sup][/sup]、IC[sup][/sup])、质谱法(GC/MS[sup][/sup]、ICP/MS[sup][/sup]、LC/MS/MS[sup][/sup])、光谱法[sup] [/sup]等。光谱法虽然操作简便,但其灵敏度不高,而[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法[sup][/sup]只能适用于水样等简单基质,用于植物源样品检测时干扰太大;用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]和[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]技术检测时,需要将PMG和APMA衍生转化为可气化物质,其引入试剂多、过程相对繁琐,效率较低[sup][/sup];用LC/MS/MS法直接检测时[sup][/sup],由于PMG和APMA分子量(分别为169、111)均较小,其主要碎片离子的质荷比多在100以下,检测实际样品时受基质干扰严重,灵敏度较低,因此柱前衍生——[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]法成为近年来国内外检测PMG和APMA残留的主流方法[sup][/sup]。以9-芴甲氧羰酰氯(FMOC-Cl)做为衍生试剂,在硼酸盐缓冲溶液中与PMG和APMA水提取液相容性好,过程简单,其衍生产物在LC/MS/MS中响应信号高,碎片离子干扰小,适合定性定量分析。 目前,采用柱前衍生——LC/MS/MS法检测茶叶、稻米等基质中PMG和APMA残留的报道很多[sup][/sup],专门针对大豆基质的报道很少。行业标准[sup][/sup]的适用范围虽然包括了大豆基质,但该方法在实验过程中试剂用量大、操作繁琐(反复调pH值)、衍生时间长(需过夜),尤其是使用阳离子交换柱(CAX)洗脱时需要加入11 mL 1%的盐酸甲醇水(20/80,v/v),水分含量过高导致旋转蒸发时很难蒸干,容易造成PMG和APMA回收率不稳定。本文专门针对大豆这类高蛋白、高脂肪含量的特殊基质,采用纯水作为提取试剂,二氯甲烷去除脂溶性杂质,C[sub]18[/sub]固相萃取小柱净化后采用FMOC-Cl衍生,最后用UPLC/MS/MS测定。该方法前处理过程简便、快速、灵敏度高,适用于大豆中PMG和APMA的残留检测。[b]1 实验部分[/b]1.1 材料与试剂 草甘膦、氨甲基膦酸(纯度≥99%,德国Dr.Ehrensorfer公司);FMOC-Cl(纯度99%,Sigma公司),使用时配置成10g/L的丙酮溶液;乙腈、二氯甲烷、甲酸、乙酸铵(色谱纯,美国Fisher公司);十水四硼酸钠(优级纯,天津市科密欧化学试剂有限公司),使用时配置成50g/L的水溶液;实验用水为Millipore纯水仪制备;C[sub]18[/sub]固相萃取小柱(200mg/3ml,美国Agilent公司)。1.2 仪器与设备 Acquity UPLC型超高效液相色谱仪(Waters公司);XEVO TQ-S三重四级杆质谱仪(Waters公司);CR21GⅢ型高速离心机(HITACHI公司);KQ5200DB型台式超声波仪(昆山市超声仪器有限公司);涡旋混合器(IKA公司)。1.3 标准溶液的配置 分别称取草甘膦和氨甲基膦酸标准品10mg(精确到0.1mg),用水溶解并定容至10mL,配置成质量浓度为1.0 mg/mL标准储备液,于4℃冰箱保存待用;使用时用水逐级稀释成所需浓度的混合标准工作液。1.4 样品前处理 提取:称取粉碎均匀后的试样1.0g(精确到0.01g)于50mL聚乙烯离心管中,加入10.0mL超纯水,涡旋混合30 s并超声提取20 min后,以10000 r/min离心3 min,将上清液转移至另一离心管中,加入5 mL二氯甲烷涡旋混合30 s,以10000 r/min离心3 min,上清液待净化。 净化:取2.5 mL上清液加入到C[sub]18[/sub]固相萃取柱(使用前依次用3mL甲醇和3mL超纯水活化)中,弃去最初的几滴流出液(约0.5 mL),将剩余部分用5 mL玻璃管收集,待衍生。 衍生:取1.0 mL净化液于5 mL离心管中,依次加入1.0 mL 50g/L的硼酸钠溶液和 1.0 mL 10g/L的FMOC-Cl衍生液,混匀后室温下衍生2 h,以10000 r/min离心3 min,取上清液过0.22 mm有机系微孔滤膜后,供UPLC/MS/MS分析测定。1.5 液相色谱及质谱条件 液相色谱:色谱柱:Waters BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm);柱温:30℃;流速:0.5 mL/min;进样量:2 μL;流动相A:乙腈;流动相B: 2 mmol /L 的乙酸铵水溶液。梯度洗脱程序:0~0.5min,10% A;0.5~3. 0 min,10%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1min,100% A~10% A,4.1 ~5.0min 10% A。 质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.2 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[align=center]表1 PMG-FMOC和 AMPA-FMOC的MRM质谱参数[/align][align=center]Table 1 MRM parameters of PMG-FMOC and AMPA-FMOC[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Cone/V[/align][/td][td][align=center]Parent ion/(m/z)[/align][/td][td][align=center]Daughter ion/(m/z) [/align][/td][td][align=center]Collision energy/V[/align][/td][/tr][tr][td][align=center]PMG-FMOC[/align][align=center] [/align][align=center]AMPA-FMOC[/align][/td][td][align=center]30[/align][align=center] [/align][align=center]30[/align][/td][td][align=center]392[/align][align=center][sup] [/sup][/align][align=center]334[/align][align=center][sup] [/sup][/align][/td][td][align=center]88[/align][align=center]214﹡[/align][align=center]112﹡[/align][align=center]179[/align][/td][td][align=center]14[/align][align=center]8[/align][align=center]11[/align][align=center]20[/align][/td][/tr][/table]﹡quantitative ion[b]2 结果与讨论[/b]2.1 色谱及质谱条件的优化 流动相的选择:对比了酸性体系(0.1%甲酸水溶液)与非酸性体系(乙酸铵水溶液)分别于甲醇、乙腈的流动相体系组合,结果发现两种分析物在酸性体系中分离效果欠佳,峰形拖尾严重,而在非酸性体系中其色谱分离效果得到明显改善,峰形对称;乙腈比甲醇体系洗脱能力更强,可以有效缩短分析时间。故本研究采用乙酸铵水溶液+乙腈流动相体系,并比较了1、2、5 mmol/L三种乙酸铵浓度与乙腈的组合,结果发现随着乙酸铵浓度的增加,目标物响应值虽略有提高但相差不大,而同时仪器背景值却显著升高,综合考虑目标物信号强度、信噪比、色谱分离效果以及分析时间等因素,本实验最终选择了2 mmol /L 乙酸铵水溶液+乙腈分析体系。质谱的选择:PMG、 AMPA对应的衍生物PMG-FMOC、AMPA-FMOC分子量分别为391、333。用超纯水配置10 mg/L 混合标准溶液直接注射到质谱中,在正负离子模式下分别进行母离子全扫描,发现正离子模式下392、334具有很好的响应,然后分别以392、334为母离子进行子离子全扫描,各得到两组丰度高、干扰小的子离子对进行MRM监测,最终确定的质谱条件见表1。2.2 前处理条件的优化 提取溶液的选择:PMG和APMA属于强极性物质,易溶于水,难溶于有机溶剂,故一般采用极性溶剂提取,如纯水及KOH、NaHCO[sub]3[/sub]溶液等[sup][/sup]。实验发现,用碱性溶液提取后,大豆中脂肪、蛋白等物质会与碱性物质发生反应,导致离心后的提取液异常浑浊,不利于后期净化和衍生,因此本实验采用纯水作为提取试剂,再经二氯甲烷液液萃取去除脂溶性杂质。 净化柱的选择:研究发现,对提取后的溶液不经SPE净化直接进行衍生, PMG和APMA的回收率均不足30%,且精密度很差,这可能是由于大豆中富含脂肪、蛋白质等物质干扰衍生过程,故本实验比较了对脂肪、蛋白质有很好去除效果的C[sub]18[/sub]、中性Al[sub]2[/sub]O[sub]3[/sub]、HLB固相萃取SPE柱的净化效果,结果发现提取液经中性Al[sub]2[/sub]O[sub]3[/sub]净化后,PMG和APMA几乎检测不到;而C[sub]18[/sub]净化后目标物回收率为92.7%、90.8%,HLB为83.6%、80.5%。故本实验选取了净化效果更好,成本相对低廉的C[sub]18[/sub]固相萃取小柱。 衍生条件的优化:FMOC-Cl的衍生机制是在碱性环境下(pH=9.0)通过FMOC-Cl基团取代目标化合物氮原子上的氢,从而生成较稳定的化合物FMOC-Cl。参照行业标准[sup][/sup]及文献报道[sup][/sup]所选用的缓冲液浓度,本实验采用50g/L的硼酸钠水溶液缓冲液体系,设置的衍生试剂质量浓度为1、2、5、10、20 g/L FMOC-Cl丙酮溶液,按照本文1.4步骤对PMG和APMA质量浓度为0.5 mg/L的纯水溶液和大豆空白基质溶液分别进行衍生,结果见图1。结果表明,在纯水溶液中,FMOC-Cl浓度为2 g/L时,PMG和APMA的峰面积已达到最大,随着衍生化试剂浓度的升高,其峰面积无明显变化;而在大豆空白基质溶液中,FMOC-Cl低浓度(1、2g/L)时,PMG和APMA几乎检测不到,其峰面积随衍生化试剂浓度增加而加大,浓度到达一定程度(10 g/L)时,峰面积不再变化。产生这种现象的原因,可能是由于尽管大豆提取液经过了二氯甲烷和C[sub]18[/sub]小柱的净化,但还是会有少量水溶性蛋白、脂肪等杂质残留在净化液中,这些杂质可能会与衍生试剂反应,影响目标物的衍生效果。研究还发现,当FMOC-Cl浓度为20 g/L时,得到的PMG和APMA色谱峰产生拖尾现象,可能是由于衍生试剂化学性质较活泼,其用量大时,过量的FMOC-Cl会迅速转化成FMOC-OH,干扰目标物峰形。在50g/L硼酸钠水溶液、10 g/L FMOC-Cl丙酮溶液条件下,考察不同时间(0.5h、1h、2h、4h、8h和16h)对衍生效果的影响,结果发现,2 h后PMG和APMA的测定值无明显增加。因此,本实验最终选定的衍生条件为50g/L硼酸钠水溶液、10 g/L FMOC-Cl丙酮溶液,室温下衍生2 h。[img=,596,378]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020904_01_2984502_3.png[/img][img=,690,530]http://ng1.17img.cn/bbsfiles/images/2017/07/201707020904_02_2984502_3.png[/img]2.3 基质效应的考察 基质效应(主要是抑制)是LC/MS/MS仪器检测时经常遇到的现象。由于本实验采用极性很强的水作为提取剂,大豆中的色素、脂肪酸等极性较强的物质也有少部分进入到最后的上机液中,在离子化带电过程中会与目标物产生竞争,抑制目标物的离子化效率。实验考察了用PMG和APMA的纯水标样去标定经过本文1.4步骤处理后的大豆空白基质溶液配置的同浓度标样,其色谱图见图2。结果发现,PMG在纯水和大豆空白基质中峰面积基本一致,而APMA在大豆空白基质中的峰面积仅为纯水中的55.7%,产生了明显的基质抑制效应。为了消除基质干扰,本实验选用大豆样品空白基质配置不同浓度的标准溶液来绘制标准曲线进行校准。2.4 线性范围和定量限 用大豆空白基质溶液分别配置0.001、0.005、0.01、0.05、0.1、0.2、0.5 mg/L的PMG和APMA混合标准溶液,按本文1.4步骤衍生后测定,以各自定量离子的峰面积为Y对应质量浓度X(mg/L)做标准曲线,得到的线性方程和相关系数见表2。结果表明,这两种物质在0.001~0.5 mg/L浓度范围内线性良好,相关系数R分别为0.9996和0.9993。以10倍信噪比(S/N)计算,该方法PMG和APMA的定量限(LOQ)均为0.01 mg/kg。[align=center]表2 PMG和APMA大豆基质标准溶液的线性方程、相关系数和定量限(LOQ)[/align][align=center]Table 2 Linear equations,correlation and LOQ of PMG and APMA in the soybean matrix standard solutions[/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Linear range/(mg/L)[/align][/td][td][align=center]Linear equation[/align][/td][td][align=center]R[/align][/td][td][align=center]LOQ/(mg/ kg )[/align][/td][/tr][tr][td][align=center]PMG[/align][align=center]AMPA[/align][/td][td][align=center][sup]0.001~0.5[/sup][/align][align=center][sup]0.001~0.5[/sup][/align][/td][td][align=center]Y=889809x+1671.3[/align][align=center]Y=476982x+1161.9[/align][/td][td][align=center]0.9996[/align][align=center]0.9993[/align][/td][td][align=center]0.01[/align][align=center]0.01[/align][/td][/tr][/table]2.5 回收率和精密度 称取大豆空白试样1.0 g,分别添加0.02、0.2、2 mg/kg水平的PMG和APMA混合标样,每个水平重复6次,按照本文1.4步骤前处理方法处理后上机检测,实验结果见表3。从表3可以看出,PMG的平均回收率为80.2%~91.5%,相对标准偏差(RSD,n=6)为3.37%~6.96%;APMA的平均回收率和RSD分别为77.7%~89.3%和4.11%~8.27%。[align=center]表3 大豆中PMG和APMA的加标回收率和相对标准偏差(n=6)[/align][align=center]Table 3 Recoveries and relative standard deviations(RSD)of PMG and APMA spiked in the soybean(n=6) [/align][table][tr][td][align=center]Analyte[/align][/td][td][align=center]Spiked level(mg/kg)[/align][/td][td][align=center]Recovery/%[/align][/td][td][align=center]RSD/%[/align][/td][/tr][tr][td]PMGAMPA[/td][td][align=center]0.02[/align][align=center]0.2[/align][align=center]2[/align][align=center]0.02[/align][align=center]0.2[/align][align=center]2[/align][/td][td][align=center]80.2[/align][align=center]91.5[/align][align=center]86.8[/align][align=center]77.7[/align][align=center]89.3[/align][align=center]85.9[/align][/td][td][align=center]6.96[/align][align=center]3.37[/align][align=center]3.95[/align][align=center]8.27[/align][align=center]4.25[/align][align=center]4.11[/align][/td][/tr][/table][b]3 结语[/b] 本文建立了超高效液相色谱-串联质谱法(UPLC/MS/MS)测定大豆中草甘膦及其代谢物氨甲基膦酸残留的分析方法。该方法灵敏度高,PMG和APMA定量限(LOQ)达到0.01 mg/kg,能满足大豆产品相关限量标准要求。同时该方法具有较高的准确度和精密度,前处理步骤简单快速,特别适合大批量大豆样品的检测。
我公司现在在做草酰氯,用气相分析含量,以前可以很好的分离出三氯化磷和三氯氧磷,但现在分不开了,且出峰时间也大幅提前。不知道为什么有哪位老兄可以指教一下啊?我的检测器是热导的,柱子SE--30在线等
如上啊,草酰氯这个产品本公司做了十几年了,但分析上一直不如意,不知道换了多少方法,就是不如意,有同类产品的朋友可以相互交流一下
谁能告诉我“对氯苯乙酰氨”是什么东西,有其他名称吗?
谁能告诉我“对氯苯乙酰氨”是什么东西,有其他名称吗?
dear,最近要开展偶氮二甲酰氨的项目,需要用LCMSMS来完成。我知道偶氮二甲酰氨高温降解,但是还是想问问,各位有谁用LCMSMS做过这个化合物呀?联二脲呢?谢谢
参照《HJ 168-2010》附录A:A.1.2中的检出限算法,做“氨-方法验证”的检出限,得到的是吸收液的检出限吗?国标上氨的曲线是用水定容的,不是吸收液定容。算出来能不能算作吸收液的检出限?国标上为0.5ug/10mL吸收液检出限。求助,谢谢。
最近公司接了一个水样,需要分析水中的氨氮含量。结果发现直接显色和蒸馏后显色差距很大,相差十倍,不知道是什么原因。觉得水中氨氮含量应该很大,因为有很重的氨味。不知道有没有哪位遇到过这种情况,到底是什么原因造成的,希望各位大侠赐教,不甚感激。
做荞麦根系分泌草酸,HPLC测定条件:0DS-C18柱,流速0.8ml/min,柱温40度,进样量20微升,分析时间15min,流动相:900ml 0.01mol/L磷酸氢二氨用磷酸调pH到5.75,再加入2ml正辛胺溶于98ml乙腈的溶液。现在做了几次,每次草酸的保留时间都不一样(11多分钟、9分钟多、8分钟多),哪位高手可以指点一下,用什么流动相测定草酸,保留时间比较稳定啊?谢谢了
各位大侠,不知道这个主题发到这里合不合适,我找了半天,不知道发在哪个版块。有CIPAC手册的大侠,帮我查查,溴丁酰草胺 CIPAC代码是多少?谢谢啦
那为大侠有草酰氯的检测标准(方法),可以给我发一份吗,谢谢啊!我的油箱sheny123@tom.com
各位大侠,不知道这个主题发到这里合不合适,我找了半天,不知道发在哪个版块。有手册的大侠,帮我查查,溴丁酰草胺 CIPAC代码是多少?谢谢啦
请问大家职业卫生里氨-纳氏试剂分光光度法的方法检出限怎么做的呢,是不能低于它的方法检出限0.2微克每毫升吗?我们做来不知道是不算的方法不对,怎么都没有那么低呢
仙鹤草的薄层鉴别,展开剂为石油醚(60-90):乙酸乙酯:醋酸=100:9:5,取上层液,可是实验中,不分层,各位什么问题?







 我要推广仪器
我要推广仪器
 下载APP
下载APP