今天做樟脑苯扎铵甲基硫酸盐原料炽灼残渣后残渣还有25%左右,但是原料标识含量为97%,请问,如何去确定残渣的含量?还是说硫酸盐类不做这方面的测定?
我们做的直读光谱,现在要求我们注意下残余元素含量,只找到了碳钢的残余元素含量标准,不锈钢和镍基材料的标准找不到。求知道的朋友分享下
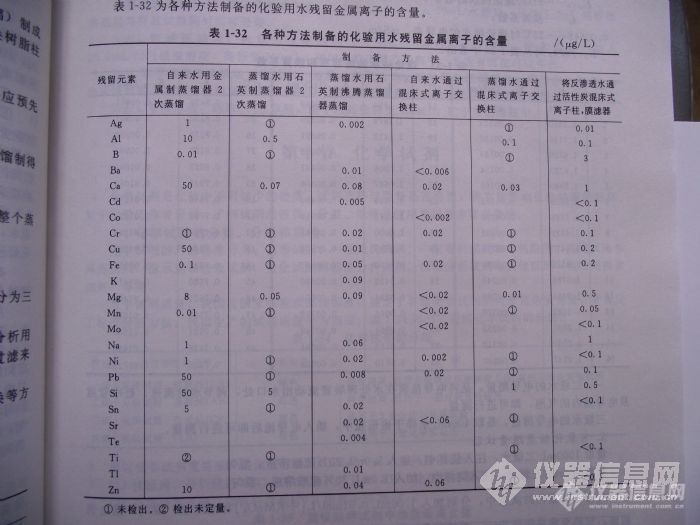
各种方法制备的化验用水残留金属离子的含量[img]http://ng1.17img.cn/bbsfiles/images/2010/05/201005231500_220281_1965589_3.jpg[/img]
http://www-bioon.qiniudn.com/bioindustry/UploadFiles/201405/2014051913423746.png我国每年进口的转基因大豆有数千万吨,主要是耐草甘膦除草剂的品种,主要用于提取食用大豆油。由于在种植期间反复喷洒草甘膦,不可避免地使转基因大豆种子中有一定的草甘膦及其次生代谢物氨甲基膦酸残留。中国医学科学院北京协和医学院放射医学研究所对阿根廷进口的转基因大豆及其制品转基因大豆油和酿制的酱油请具有农药残留分析检测资质的第三方检测机构的分析测试表明,进口的转基因大豆农残含量分别为:草甘膦3.908mg/kg和氨甲基膦酸3.364mg/kg。我国进口的转基因大豆的农残含量较高。
求助一份 "ISO 8974 -2002 塑料.酚醛树脂.用气体色谱法测定残余酚的含量"标准,有请给一份!
合成树脂乳液中残余单体含量的气相色谱测定方法探讨 吴亚虎,韩婷婷(广东中山市巴德士化工有限公司品控中心,528427) 摘要:讨论了用气相色谱内标法测定合成树脂乳液中未反应完的残余单体含量的方法,并对该方法的精密度、准确度进行了考察/实验表明,该方法简单易行,准确可靠,适用于各种合成树脂乳液样品。 关键词:气相色谱;极性小口径毛细管柱;内标法; 醋酸乙烯酯、甲基丙烯酸甲酯、苯乙烯、丙烯酸丁酯、丙烯酸异辛酯等单体。 0.引言 自国家十项强制性标准颁布以后,市面上各种涂料中的有害物质必须达到国家相关限量标准。由于合成树脂乳液中未反应的残余单体对人的身体健康和环境会带来不同程度的影响,为此必须设法控制乳液中残余单体的浓度……..目前国科多采用顶空进样技术分析乳液中的残余单体含量,由于仪器投资较大,且样品回收率也不理想,样品前处理较烦锁,对于中小企业这样投资较少,易于在中小企业中推广,该分析方法较难于推广,而我们介绍的是普通分析方法。采用小口径毛细管柱,用氢火焰离子化检测器(FID)进行检测,以内标法定量。柱温采用程序升温,分离效果十分理想。其加标回收率分别在94%-103%之间,分析结果的精密度、准确度完全达到检测要求。 1.实验部分: 1.1 仪器和试剂 电子分析天平(万分之一);GC5890F气相色谱仪(带有分流装置);1.0ul微量进样器;20ml带胶塞小玻璃瓶若干;医用注射器1ml、2ml各两只;小口径毛细柱DB-17HT(0.25mm x 30m x 0.15um;最高使用温度为360℃);积分仪或色谱工作站。醋酸乙烯酯VAM(色谱纯)、甲基丙烯酸甲酯MMA(色谱纯)、苯乙烯ST(色谱纯)、丙烯酸丁酯BA(色谱纯)丙烯异辛酯2-EHA(色谱纯)、丙酮(分析纯)配成4+1混合水溶液(作稀释剂),水(纯净水或蒸馏水)。内标物:环已酮(色谱纯) 1.2 测定原理试样中加适量内标物并用少许丙酮(4+1)稀释摇匀后,用微量注射器将稀释后的溶液注入气相色谱仪,样品被载气带入色谱柱,在柱内被分离成相应的组份,用氢火焰离子化检测器检测并记录色谱图,反数据用内标法计算试样溶液中各待测残余单体的含量。 1.3 测定条件 气化温度:280℃ 检测温度:320℃ 载气:氮气:纯度≥99.99%,变色硅胶+5A分子筛除水、除油,柱前压力为60Kpa(30℃); 氢气:纯度≥99.99%,变色硅胶+5A分子筛除水、除油,柱前压力为65Kpa(30℃); 空气:变色硅胶[/fon
谁能告诉我GB/T4615-2008 CRVCM=As/Rf(4.257768×10-3+6.095721×10-2/W)中4.257768×10-3和6.095721×10-2是怎么来的,谢谢!固上气相色谱法测定聚氯乙烯树脂中 残留氯乙烯单体含量(参考件) A.1 氯乙烯标准气和标准样的配制A.1.1 标准气的配制 从耐压氯乙烯容器中,用注射器取出5ml氯乙烯气体,注入已密封的样品瓶中,其浓度C2(ml/ml)按式(A1)计算: C2=V2/(V1+V2) 式中:V1——样品瓶的体积,23.5ml ; V2——加入氯乙烯气体的体积,ml 。A.1.2 标准样的配制 在两个系列各三个样品瓶中,用微量注射器,分别准确地注入标准气1.2、12和120ul。每个标准样中氯乙烯单体(VCM)含量(ug/ml)按式(A2)计算: VCM= C2×V/V3×106 式中:C2——标准气中氯乙烯的体积浓度,ml/ml ; V——加入的标准气的体积,ml ; V3——样品瓶的体积,ml 。 注:如果能预先估计被测试样中氯乙烯含量,可只配一个含量与被测试样中含量接近的标准样,而不必同时做三个标准样。A.2分析步骤A.2.1 试样的制备 称取已混合均匀的树脂样品4±0.5g(精确到0.1mg),置于样品瓶中,并立即盖紧。A.2.2 样品的平衡 将标准样和试样瓶一起置于恒温器中(90±1℃),恒温60min以上,使氯乙烯在气固两相中达到平衡。取出1ml上部气体注入色谱仪。A.3 结果表示A.3.1 试样中残留氯乙烯单体(RVCM)含量(mg/kg)按式(A3)计算: RVCM=As/Rf(4.257768×10-3+6.095721×10-2/W) 式中:As——试样中RVCM峰面积,cm2 ; Rf——响应因子(标样峰面积/标样体积,cm2/PPm); W——试样质量,g 。A.3.2 采用本方法时的试验条件如下: 室温:22℃(295K); 平衡温度:90℃(363K); 大气压力:750mmHg ; 样品瓶体积:23.5ml ; 试样含水量:低于0.5% 。
樟脑苯扎铵甲基硫酸盐原料炽灼残渣后残渣还有25%左右,但是原料标识含量为97%,请问,如何去确定残渣的含量?是不是在残渣中有些硫酸盐类存在,如何去扣除呢?
据说日本有专门的书折算脱水蔬菜农残含量与新鲜蔬菜农残含量的相当值。我国似乎没有这方面的资料。各位有相关资料推荐吗?谢谢
摘要:讨论了用气相色谱内标法测定合成树脂乳液中未反应完的残余单体含量的方法,并对该方法的精密度、准确度进行了考察/实验表明,该方法简单易行,准确可靠,适用于各种合成树脂乳液样品。 0.引言 自国家十项强制性标准颁布以后,市面上各种涂料中的有害物质必须达到国家相关限量标准。由于合成树脂乳液中未反应的残余单体对人的身体健康和环境会带来不同程度的影响,为此必须设法控制乳液中残余单体的浓度……..目前国科多采用顶空进样技术分析乳液中的残余单体含量,由于仪器投资较大,且样品回收率也不理想,样品前处理较烦锁,对于中小企业这样投资较少,易于在中小企业中推广,该分析方法较难于推广,而我们介绍的是普通分析方法。采用小口径毛细管柱,用氢火焰离子化检测器(FID)进行检测,以内标法定量。柱温采用程序升温,分离效果十分理想。其加标回收率分别在94%-103%之间,分析结果的精密度、准确度完全达到检测要求。 1.1实验部分:1.1仪器和试剂电子分析天平(万分之一);GC7800气相色谱仪(带有分流装置);1.0ul微量进样器;20ml带胶塞小玻璃瓶若干;医用注射器1ml、2ml各两只;小口径毛细柱DB-17HT(0.25mmx30mx0.15um;最高使用温度为360℃);积分仪或色谱工作站。醋酸乙烯酯VAM(色谱纯)、甲基丙烯酸甲酯MMA(色谱纯)、苯乙烯ST(色谱纯)、丙烯酸丁酯BA(色谱纯)丙烯异辛酯2-EHA(色谱纯)、丙酮(分析纯)配成4 1混合水溶液(作稀释剂),水(纯净水或蒸馏水)。内标物:环已酮(色谱纯) 1.2测定原理 试样中加适量内标物并用少许丙酮(4 1)稀释摇匀后,用微量注射器将稀释后的溶液注入气相色谱仪,样品被载气带入色谱柱,在柱内被分离成相应的组份,用氢火焰离子化检测器检测并记录色谱图,反数据用内标法计算试样溶液中各待测残余单体的含量。 1.3测定条件以样品中各组分是否完全分离开为依据而确认测定条件: 气化温度:280℃检测温度:320℃载气:氮气:纯度≥99.99%,变色硅胶 5A分子筛除水、除油,柱前压力为60Kpa(30℃);氢气:纯度≥99.99%,变色硅胶 5A分子筛除水、除油,柱前压力为65Kpa(30℃);空气:变色硅胶 5A分子筛除水、除油,柱前压力为55Kpa(30℃);柱温采用程序升温:初温30℃,恒温3min,以10℃/mm升温速率升至140℃,再以50℃/min升温速率升至260℃保持4min.分流比:45:1进样量:0.2ul1.4相对校正因子的测定1.4.1标准样品的配制于20ml样品瓶中分别称取0.03g(精确至0.0001g)的醋梭乙烯酯VAM、丙烯酸丁酯Bt等待测单体的色谱纯品和内标物环已酮,加入纺2mL丙酮(4 1)稀释,用力摇匀3min.(注意:每次衡量后应立即将样品瓶盖紧,以防止样品挥发损失。) 1.4相对校正因子的测定待仪器稳定后,吸取0.2uL标准样品注入色谱仪,记录色谱图和色谱数据……。典型合成树脂乳液的色谱图各单体及内标物的相对保留时间分别是:VAM1.771’;MMA3.078’;BA5.498’;St5.683’;2-EHA8.495’;内标环已酮6.218’1.4.3相对校正因子的计算醋酸乙烯酯、丙烯酸丁酯、苯乙烯等各需待测的残余单体对环已酮的相对校正因子Fi按下式计算:式中:mi——待测残余单体各自的质量;gAs——内标物环已酮的峰面积;ms——内标物环已酮的质量;gAi——待没残余单体各自的峰面积。 1.5连续平行测得待测残余单体各自对环已酮(内标物)的相对校正因子Fi的平等偏差应小于0.05.1.5加标回收率试验为了证明测定结果的可靠性,称取一定量的各待测单体纯品配制一已知尝试溶液,按上述分析方法测定各溶液的浓度,计算各自的回收率,结果见表(1) 1.6样品的测定将样品搅拌均匀后,在20mL样品瓶中准确称取2g左右和一小滴(用1mL注射器)内标物(约0.001g)环已酮于样品瓶中,加入适量(2~3mL)的丙酮(4 1)稀释剂,立即加盖瓶塞,充分摇匀2min.在相同于测定校正因子的分析条件下,用1ul微量进样器取0.2ul该试液注入色谱仪,并记录色谱图和数据,根据待测残余单体各自对内标物的保留时间进行定性。 1.7结果的计算待测残余单体各自的质量分数按下式计算:式中:Fi——待没单体各自对内标物的相对校正因子;Ms——内标物的质量,gAi——试样中待测残余单体各自的峰存积;Mi——待没试样的质量,gAs——内标物的峰面积取平行测定2次结果的算术平均值作为试样中各待测单体的测定结果。 1.8重现性同一操作作者两次测定结果的相对偏差小于0.5%2.结果与讨论(1)该分析方法选用中等极性小口径耐高温的毛细柱,进样量不能大于0.2ul,且分流比要足够大。由于各残余单体的沸点相对较高,所以我们选用耐高温的毛细柱,且该柱对酯类有较佳的分离效果。 (2)汽化室内须配有石英玻璃衬管,内填适量经处理的玻璃棉,以防止残留物进入毛细柱,并要勤换衬管为妥。 (3)因各单体对内标物的相对响应值不同,的以在配制标准溶液时,称取的各单体质量不一定都是0.03g,原则是尽量使各单体的峰面积和内标物的峰面积之比值接近于1。 (4)某些样品经丙酮稀释旋转一段时间后会出现破乳分层,做样品时须取其上层清液进色谱仪分析。 (5)醋梭乙烯酯VAM存在水的现象。因此在做其回收率时溶剂用脱水分析砘丙酮这样能有效控制VAM水解。但测试样时,溶剂为丙酮水溶液(4 1),这是由于有些样品溶于丙酮(脱水)时会产生胶化现象。
意思就是车间做完一个产品后 进入到下一个程序 来验证下该仪器是否已经清洗干净 我们需要测下残留物含量(紫外) 大概怎么测呢 你们是怎么考察的?
[color=#444444]产品中残留有少量三溴苯酚钠,与空气氧化成红色;怎样才能检测出三溴苯酚钠的残留量呢?[/color][color=#444444]之前的一个思路:将产品用二氯甲烷溶解,加盐酸酸化,三溴苯酚钠转化成三溴苯酚,再用液相色谱分析;但是出峰很小,非正常产品含量0.06%左右,正常产品含量0.01%,但是后期再用相同的方法检测,三溴苯酚不出峰了,感觉很奇怪,请大神解答。。。[/color]
我做顶空有机残留用的是外标法,根据计算公式一直都认为求得的数据是含量,可是一个同行(比较有经验一点)说求得的是纯度不能和含量混为一谈,我晕了,想和各位色友讨论一下。
求GB/T 8293-2008 浓缩天然胶乳 残渣含量的测定
樟脑苯扎铵甲基硫酸盐原料炽灼残渣后残渣还有25%左右,但是原料标识含量为97%,请问,如何去确定残渣的含量?
求助乙氧氟草醚检测,是主含量检测不是残留,谢谢
求上述新标准,谢谢了。GB/T 4615-2008 聚氯乙烯树脂 残留氯乙烯单体含量的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[em0808]
聚氯乙烯树脂残留氯乙烯单体含量的测定 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 GB-T 4615-2008[~123634~]
日本厚生劳动省发布食安输发0624第4号通报:近日,根据2014年3月28日发布的食安输发0328第10号通报(于2014年6月13日修正为食安输发0613第4号通报),通过对相关产品所采取的强化监视检查的情况来看,将取消对于中国产养殖鳗鱼加工品(仅限于烤鳗鱼以及以鳗鱼为主料的其他食品)中恩诺沙星残留含量的强化监视检查。日本厚生劳动省在通报中称,今后对于中国生产的上述产品相关项目的残留含量检查项目将依照通常的监视检查体制进行检查。对于实行自主检查的进口商,仍然按照原有的检查体制进行。
有的脂肪含量为什么是用酸度衡量的啊?我想要鱼肉香肠和酸奶的脂肪含量国标
求助:残留在水溶液中脂肪胺比如十二胺,可以[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析含量吗?有大神测过吗?请分享经验,谢谢!
各位大侠 帮忙! 标准 YBB00102002 药用聚酯瓶的测乙醛残留含量 不得超过千分之二 如何计算?是对照品和样品的峰面积之比吗?
上周发了帖子,不知道为什么没有出来。十多年没发帖子了,本来不想发,但觉得还是有必要交流一下。根据GB 2763中的附录A 表A. 1中要求,核果类水果及部分热带水果,去核检测的样品,残留量应计入果核的重量。下面用个例子说明一下。某批次桃样品去除柄部后称重500g(含果核),收集果核称重100g。将去除柄和核的样品粉碎均匀,检测结果为0.1mg/kg,计算最终样品中农残含量为多少?首先,要明白两个前提,一是果核中有没有农药残留?是多少?二是要搞清楚一个逻辑,计入果核的重量后,含量是增加还是减少了。第一个,显然去除果核,也是默认果核中没有农药残留,或者不影响食用安全;第二个,既然果核中没有农药残留,那计算残留量的时候计入果核的重量,最终样品的农药残留含量是降低的。明白这个逻辑很重要,也就清楚了为什么那个帖子提到的除以果肉率是错误的。那下一个问题,为什么那样算会错,而且好多同仁都认为很合理。是因为先算出来一个果肉比,造成了误导。除以果肉比,只是得出了一个按那个比例放大了的浓度外,没有其他意义。计入果核重量,跟果肉比没关系。回到例题,先算出[size=14px]果肉[/size][size=14px]质量=[/size][size=14px]500[/size][size=14px]g[/size][size=14px]-100[/size][size=14px]g[/size][size=14px]=[/size][size=14px]400[/size][size=14px]g;[/size]则计入果核样品中农残含量=(果肉检测结果0.1mg/kg [font='times new roman'][size=16px]×[/size][/font][size=14px] [/size][size=14px]果肉[/size][size=14px]4[/size][size=14px]0[/size][size=14px]0[/size][size=14px]g[/size][size=14px])[/size]/ 500g=0.08 mg/kg。那么你会看到神奇的一幕出现了,结果竟然等于“果肉检测含量 [font='times new roman'][size=16px]×[/size][/font][size=14px] [/size][size=14px]果肉比[/size]”[size=14px]。[/size][size=14px]这也是为什么大多数人会理解有误的原因,因为本来很简单一个事,就因为先算了一个果肉比,[/size]果肉检测含量[size=14px]X[/size][size=14px] [/size][size=14px]果肉比[/size][size=14px] 理解不通啊,所以就除以果肉比吧。[/size][size=14px]如果真要用果肉比来算的话也可以,但不是用[/size]果肉检测含量除以果肉比,因为果肉比是样品质量比,应该是除在分母“0.1mg/(kg/果肉比) ”,这样就好理解了。总之计算公式是:全果样品农药残留含量mg/kg =(果肉检测结果mg/kg [font='times new roman'][size=16px]×[/size][/font][size=14px] [/size][size=14px]果肉[/size][size=14px]样品量k[/size][size=14px]g[/size])/ 全果样品量kg[size=14px]。[/size][size=14px]个人观点,仅供参考,欢迎交流讨论。[/size]
聚合物在合成出来后处理的时候要进行洗涤、过滤,但是再怎么洗也会有溶剂包附在聚合物链中,没办法彻底洗干净,我们就想准确检测这个残留溶剂的含量,是一种聚酰亚胺,含有N,N-二甲基乙酰胺(DMAc),请问各位高手,可否用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]连用给与解决呢?不能定量的话可否定性呢?
GB 4615—87代替GB 4615—84聚氯乙烯树脂中残留氯乙烯单体含量测定方法1.氯乙烯标准气和标准样的配制1.1标准气的配制:在样品瓶中几颗玻璃珠后,盖紧密封,在分析天平上称重(精确到0.1mg)用注射器从氯乙烯容器中取出5ml气体(取气时注射器先用氯乙烯气体洗两次)注入瓶中,在2称量(精确到0.1mg),摇匀后静止10min,立即使用.该气体浓度C1约为400ug/ml.可按(1)计算:C1=(W2-W1)/(V1+V2)*106 式中:W1—放进玻璃珠的样品瓶质量,g W2—放进玻璃珠的样品瓶注入了5ml氯乙烯气体后的质量,g V1—样品瓶的体积,ml V2—加入氯乙烯的体积,ml1.2标准样的配制:在两个系列各三个样品瓶中,用微量注射器分别准确地注入3mlDMAC(N,N-二甲基乙酰胺),再分别准确地注入0.5,5,50ul标准气摇匀待用。每个标准样中氯乙烯单体(VCM)的含量(ug),按式(2)计算:VCM=C1*V式中:C1—标准气浓度,ug/ml V—加入的标准气的体积,ml问题:在配标准气的时候一切正常,但稀释到标准样的时候,进样出的峰,峰高和峰面积都达不到做为样品参照的要求!请问有那位做过这方面的分析,请帮我分析一下是那步出了问题?拜托拉!!!!!!!!
REACH法规中有针对聚合物单体注册的相关内容。请问聚合物中单体含量怎么检测?包括已反应单体和残留单体。我要检测的聚合物包含好几种聚合物成分,假设没有残余单体,是不是每种聚合物的成分含量就是构成该聚合物的单体单元含量?举例ABS树脂,其中A,B,S各个组分的单体含量如何检测?
请教一下各位:果汁含量与固形物含量有什么区别,两者是一个意思吗?
索氏抽提法:轻松检测食品中的脂肪含量 给大家分享一个实用的检测方法——索氏抽提法,教你如何轻松检测食品中的脂肪含量。 1. 样品准备 首先,我们要准备好待测的样品。如果是固体,像坚果、面粉这类,直接称取2-5克;如果是液体或半固体,比如牛奶、奶酪,就要称取5-10克。样品称取好后,放入滤纸筒中,滤纸筒的高度不能超过虹吸管,否则会影响提取效果。 2. 提取器的清洗 将索氏提取器各部位充分洗涤,并用蒸馏水清洗后烘干。脂肪烧瓶要在103℃±2℃的烘箱内干燥至恒重,前后两次称量差不超过2mg。 3. 样品测定 将滤纸筒放入索氏提取器的抽提筒内,连接已干燥至恒重的脂肪烧瓶。从冷凝管上端加入乙醚或石油醚至瓶内容积的2/3处,通入冷凝水,将底瓶浸没在水浴中加热。注意,用一小团脱脂棉轻轻塞入冷凝管上口,防止空气水分和杂质进入。 4. 抽提温度与时间 水浴温度应控制在使提取液每6-8分钟回流一次,一般样品提取6-12小时,坚果样品提取约16小时。提取结束时,用毛玻璃板接取一滴提取液,如无油斑则表明提取完毕。 5. 溶剂回收与恒重 取下脂肪烧瓶,回收乙醚或石油醚。待烧瓶内溶剂仅剩下1-2mL时,在水浴上赶尽残留的溶剂。然后在95-105℃下干燥2小时,置于干燥器中冷却至室温,称量。继续干燥30分钟后冷却称量,反复干燥至恒重(前后两次称量差不超过2mg)。 故障排除小贴士 提取不完全:如果提取液仍有油斑,说明脂肪未被完全提取,需要延长提取时间。 称量不准确:干燥后的脂肪烧瓶容易吸湿,称量时要迅速,避免影响结果。 溶剂残留:确保溶剂完全挥净后再放入烘箱,否则会有爆炸危险。 安全问题:乙醚是易燃易爆物质,使用时要注意通风,避免明火。 通过索氏抽提法,我们可以快速准确地检测食品中的脂肪含量,操作简单,结果可靠。希望能对各位朋友有帮助。
最近要测定原辅料磷酸三丁酯(TNBP)的含量,可我不知道怎么测定,看到药典上用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]做的,但我又从来没做过[url=https://insevent.instrument.com.cn/t/Mp]气相[/url],所以也不清楚,而且药典上的步骤是测样品中的TNBP残余量的,这个原辅料的TNBP含量又很高(〉98%),我不知道该怎么测定,请哪位高人指点一下,给个好方法把,谢谢了。
我配嘧菌酯样品是用曝气后的自来水配制的,上机前就只过了滤膜,测出来实际含量只有理论值的一半是怎么回事呀?标液测出来的实际含量是理论含量的89-107%







 我要推广仪器
我要推广仪器
 下载APP
下载APP