我用RTXWAX柱子做甘油含量,做完后有个残留峰在10分钟左右,有什么办法把 残留峰去掉?我这跟柱子最高温度250°,甘油沸点290°,我进样口温度设的240,检测器250°,是不是不可以设高于250°啊?关键怎么把柱子的残留峰去掉?我用乙醇,丙酮,二氯乙烷试了,没有用~!
意思就是车间做完一个产品后 进入到下一个程序 来验证下该仪器是否已经清洗干净 我们需要测下残留物含量(紫外) 大概怎么测呢 你们是怎么考察的?
[color=#444444]产品中残留有少量三溴苯酚钠,与空气氧化成红色;怎样才能检测出三溴苯酚钠的残留量呢?[/color][color=#444444]之前的一个思路:将产品用二氯甲烷溶解,加盐酸酸化,三溴苯酚钠转化成三溴苯酚,再用液相色谱分析;但是出峰很小,非正常产品含量0.06%左右,正常产品含量0.01%,但是后期再用相同的方法检测,三溴苯酚不出峰了,感觉很奇怪,请大神解答。。。[/color]
求助乙氧氟草醚检测,是主含量检测不是残留,谢谢
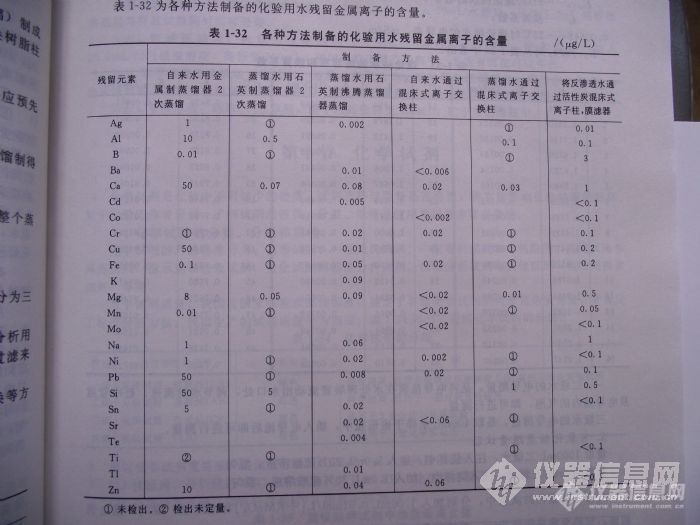
各种方法制备的化验用水残留金属离子的含量[img]http://ng1.17img.cn/bbsfiles/images/2010/05/201005231500_220281_1965589_3.jpg[/img]
上周发了帖子,不知道为什么没有出来。十多年没发帖子了,本来不想发,但觉得还是有必要交流一下。根据GB 2763中的附录A 表A. 1中要求,核果类水果及部分热带水果,去核检测的样品,残留量应计入果核的重量。下面用个例子说明一下。某批次桃样品去除柄部后称重500g(含果核),收集果核称重100g。将去除柄和核的样品粉碎均匀,检测结果为0.1mg/kg,计算最终样品中农残含量为多少?首先,要明白两个前提,一是果核中有没有农药残留?是多少?二是要搞清楚一个逻辑,计入果核的重量后,含量是增加还是减少了。第一个,显然去除果核,也是默认果核中没有农药残留,或者不影响食用安全;第二个,既然果核中没有农药残留,那计算残留量的时候计入果核的重量,最终样品的农药残留含量是降低的。明白这个逻辑很重要,也就清楚了为什么那个帖子提到的除以果肉率是错误的。那下一个问题,为什么那样算会错,而且好多同仁都认为很合理。是因为先算出来一个果肉比,造成了误导。除以果肉比,只是得出了一个按那个比例放大了的浓度外,没有其他意义。计入果核重量,跟果肉比没关系。回到例题,先算出[size=14px]果肉[/size][size=14px]质量=[/size][size=14px]500[/size][size=14px]g[/size][size=14px]-100[/size][size=14px]g[/size][size=14px]=[/size][size=14px]400[/size][size=14px]g;[/size]则计入果核样品中农残含量=(果肉检测结果0.1mg/kg [font='times new roman'][size=16px]×[/size][/font][size=14px] [/size][size=14px]果肉[/size][size=14px]4[/size][size=14px]0[/size][size=14px]0[/size][size=14px]g[/size][size=14px])[/size]/ 500g=0.08 mg/kg。那么你会看到神奇的一幕出现了,结果竟然等于“果肉检测含量 [font='times new roman'][size=16px]×[/size][/font][size=14px] [/size][size=14px]果肉比[/size]”[size=14px]。[/size][size=14px]这也是为什么大多数人会理解有误的原因,因为本来很简单一个事,就因为先算了一个果肉比,[/size]果肉检测含量[size=14px]X[/size][size=14px] [/size][size=14px]果肉比[/size][size=14px] 理解不通啊,所以就除以果肉比吧。[/size][size=14px]如果真要用果肉比来算的话也可以,但不是用[/size]果肉检测含量除以果肉比,因为果肉比是样品质量比,应该是除在分母“0.1mg/(kg/果肉比) ”,这样就好理解了。总之计算公式是:全果样品农药残留含量mg/kg =(果肉检测结果mg/kg [font='times new roman'][size=16px]×[/size][/font][size=14px] [/size][size=14px]果肉[/size][size=14px]样品量k[/size][size=14px]g[/size])/ 全果样品量kg[size=14px]。[/size][size=14px]个人观点,仅供参考,欢迎交流讨论。[/size]
最近同事在做芝麻油的溶剂残留,明明是压榨工艺,溶剂残留应该不会高,可是做的几个样含量都比较高挺奇怪的,不知道有没有版友做过这个项目,是否是芝麻油中本身有什么干扰呀?
大家都知道浸出工艺生产的植物油,是需要溶剂提取的,难免会有溶剂残留在其中。可是根据压榨工艺的描述应该不会有溶剂残留的存在,可是试剂情况却是有些压榨工艺的产品有溶剂残留甚至还出现含量较高的情况。并且在一些标准中对低等级的压榨工艺的产品的规定了一定的限量,却不是不得检出。不知道压榨工艺的植物油中溶剂残留的来源都有哪些呢?敬请各位版友发表一下自己的观点,谢谢!
[em0912]EVA中残留VA monomer的含量检测方法?
GB/T 8570.4-2010 液体无水氨的测定方法 第4部分:残留物含量 容量法
GB/T 8570.3-2010 液体无水氨的测定方法 第3部分:残留物含量 重量
用顶空测酒精中甲醇含量时,怎样消除仪器中的甲醇残留对检测结果的影响,在进了多次空白的情况下,仪器中还是会有0.3~0.4峰面积的甲醇,对于酒精中含量较低的甲醇检测,对其结果影响较大,想请问怎样处理这个问题?
测包材溶剂残留总量和苯类含量用什么柱子,很急,先谢谢了!
公司产品需要检测原料药中三苯基膦氯化铑的残留含量,希望有人告知哪里有检测的,最好有相关的资质。谢谢。
各位大侠 帮忙! 标准 YBB00102002 药用聚酯瓶的测乙醛残留含量 不得超过千分之二 如何计算?是对照品和样品的峰面积之比吗?
公司现在需要用GC检测一个聚合物中残留苯的含量,仪器也没有顶空进样器,请教各位大侠用什么方法检测啊,最好能够详细些,谢谢!
大家好,我从来没有接触过XRD,最近老板让我检测一种奥氏体不锈钢中残留铁素体的含量,我不知道怎么做才好。有的人说将试样腐蚀后,显示出残留铁素体,然后采用定量金相的方法,可是我觉得不准,还有人说采用一种磁性检测方法,前几天一个老师说,采用XRD可以做到,我不太清楚,特来请教。如果知道的大虾,多多指教,另外顺便问一下,如果做XRD,如何制样啊,我的就是一个2mm厚的不锈钢板,谢谢了[em11]
客户要求塑料中氯乙烯单体的含量要小于5ppm。那如果是PVC线皮,其中残留的氯乙烯单体的含量能够做到5ppm以下吗?有意见认为PVC材料一定做不到,因为[CH2CHCl]n里面这个n是不确定的,所以PVC是混合物。所以PVC混合物中含有氯乙烯是非常正常的。而另一方面,很多其他行业对PVC中残留氯乙烯单体的含量限值更低(比如食品接触材料——PVC保鲜膜里要求是小于1ppm),这样看来,5ppm这个限值又应该是可以做到的。电子产品中的PVC材料的生产工艺和其他行业里PVC的生产工艺是否一样呢?5ppm这个限值到底能不能做得到?
谁能告诉我GB/T4615-2008 CRVCM=As/Rf(4.257768×10-3+6.095721×10-2/W)中4.257768×10-3和6.095721×10-2是怎么来的,谢谢!固上气相色谱法测定聚氯乙烯树脂中 残留氯乙烯单体含量(参考件) A.1 氯乙烯标准气和标准样的配制A.1.1 标准气的配制 从耐压氯乙烯容器中,用注射器取出5ml氯乙烯气体,注入已密封的样品瓶中,其浓度C2(ml/ml)按式(A1)计算: C2=V2/(V1+V2) 式中:V1——样品瓶的体积,23.5ml ; V2——加入氯乙烯气体的体积,ml 。A.1.2 标准样的配制 在两个系列各三个样品瓶中,用微量注射器,分别准确地注入标准气1.2、12和120ul。每个标准样中氯乙烯单体(VCM)含量(ug/ml)按式(A2)计算: VCM= C2×V/V3×106 式中:C2——标准气中氯乙烯的体积浓度,ml/ml ; V——加入的标准气的体积,ml ; V3——样品瓶的体积,ml 。 注:如果能预先估计被测试样中氯乙烯含量,可只配一个含量与被测试样中含量接近的标准样,而不必同时做三个标准样。A.2分析步骤A.2.1 试样的制备 称取已混合均匀的树脂样品4±0.5g(精确到0.1mg),置于样品瓶中,并立即盖紧。A.2.2 样品的平衡 将标准样和试样瓶一起置于恒温器中(90±1℃),恒温60min以上,使氯乙烯在气固两相中达到平衡。取出1ml上部气体注入色谱仪。A.3 结果表示A.3.1 试样中残留氯乙烯单体(RVCM)含量(mg/kg)按式(A3)计算: RVCM=As/Rf(4.257768×10-3+6.095721×10-2/W) 式中:As——试样中RVCM峰面积,cm2 ; Rf——响应因子(标样峰面积/标样体积,cm2/PPm); W——试样质量,g 。A.3.2 采用本方法时的试验条件如下: 室温:22℃(295K); 平衡温度:90℃(363K); 大气压力:750mmHg ; 样品瓶体积:23.5ml ; 试样含水量:低于0.5% 。
聚合物在合成出来后处理的时候要进行洗涤、过滤,但是再怎么洗也会有溶剂包附在聚合物链中,没办法彻底洗干净,我们就想准确检测这个残留溶剂的含量,是一种聚酰亚胺,含有N,N-二甲基乙酰胺(DMAc),请问各位高手,可否用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]连用给与解决呢?不能定量的话可否定性呢?
[color=#444444]我最近在进行检测辅料醋酸铵里残留溶剂醋酸含量的HPLC方法学建立。结果发现醋酸含量高达78%,觉得在过程中醋酸铵转化成了醋酸,方法失败。我觉得与酸性流动相还有柱子加热到50度有关。[/color][color=#444444]流动相:0.005N硫酸水溶液(觉得有很大问题!)[/color][color=#444444]色谱柱: Amiex;柱温:50度(?不太确定有无影响)[/color][color=#444444]溶剂:乙腈 [/color][color=#444444]希望哪位做过相关方法学可以给些建议,特别是在流动相的选择上![/color]
我做的是香精香料的检测,有时候那些样品都需要标定残留苯或甲苯的含量,通常这两个的含量都很微量,谁有定性和定量的方法,请赐教
http://www-bioon.qiniudn.com/bioindustry/UploadFiles/201405/2014051913423746.png我国每年进口的转基因大豆有数千万吨,主要是耐草甘膦除草剂的品种,主要用于提取食用大豆油。由于在种植期间反复喷洒草甘膦,不可避免地使转基因大豆种子中有一定的草甘膦及其次生代谢物氨甲基膦酸残留。中国医学科学院北京协和医学院放射医学研究所对阿根廷进口的转基因大豆及其制品转基因大豆油和酿制的酱油请具有农药残留分析检测资质的第三方检测机构的分析测试表明,进口的转基因大豆农残含量分别为:草甘膦3.908mg/kg和氨甲基膦酸3.364mg/kg。我国进口的转基因大豆的农残含量较高。
农药残留检测中经常会碰到含油植物样品,而油会成为目标物的共萃物影响上机分析,看贴子上有说用过很多种SPE柱都能除油,大家能说说自己用过的前处理方法中用到的SPE柱的除油方法和除油效果吗?[color=#DC143C][size=4]欢迎版友把自己用过的经验拿出来大家分享,有奖励.[/size][/color]
GB/T 8570.4-2010 液体无水氨的测定方法 第4部分:残留物含量 容量法
GB/T 8570.3-2010 液体无水氨的测定方法 第3部分:残留物含量 重量法
背景介绍某一药物中间体在制备过程中使用了异丙硫醇,需要对异丙硫醇的含量做相应质控要求。因该中间体沸点高不易气化且异丙硫醇的含量可能较低,故选择顶空进样来测定其含量。资料调研调研文献资料并未查到有已知的测定残留异丙硫醇含量的方法,故需要摸索条件建立检测方法。先看目标物“异丙硫醇”的物化特性:分子式: C3H8S 化学结构式:http://ng1.17img.cn/bbsfiles/images/2014/12/201412291106_529791_2167114_3.bmp 分子量: 76.15CAS No.: 75-33-2密度:0.823g/cm3熔点:-131℃沸点:55.8°Cat760mmHg溶解性:微溶于水,溶于乙醇、乙醚等。根据异丙硫醇的物化特性,气相色谱柱可选择中强极性的柱子,顶空进样可选择水作为溶剂。经过一系列的摸索最终确立检测方法如下:方法建立1. 仪器:PE Clarus 680+SQ8 GCMS;TurbomMatrix 40 HS进样器2. 色谱柱:Agilent DB-624,30m*0.250mm,1.40um3. 升温程序:40℃ Hold 6min;以15℃/min的速率升至220℃,Hold 6min。4. 顶空参数设置: 炉温60℃并跟踪炉温(即针温65℃,传输线70℃);捕集肼温度:Hi-300℃,Lo-39℃; 捕集肼保持4min,炉保温10min,干吹2min,解吸0.5min; 色谱柱压力16.0psi,瓶压40psi,解析压力5.5psi;捕集肼1个循环。5. MS参数:EI+源,源温度240℃,传输线250℃ 0.00-4.00min Solvent delay 4.00-24.00min Scan Mass “50-200”样品及标品配置1. 空白对照:准确移取5ml纯化水于顶空进样瓶中,上机待测;2. 样品:0.1487g+5ml纯化水于顶空进样瓶中,上机待测;3. 标准储备液的配置:标品信息名称:2-丙硫醇 CAS No.: 75-33-2供应商:北京百灵威科技有限公司Lot:LHC0N23含量:98%标准储备液的配置:0.0193g标品于100ml容量瓶中,纯化水溶解后超声定容,待用。Std1:准确移取2000ul储备液至5ml容量瓶中,纯化水定容后转移至顶空进样瓶中,上机待测.Std2:准确移取200ul储备液至5ml容量瓶中,纯化水定容后转移至顶空进样瓶中,上机待测;Std3:准确移取40ul储备液至10 ml容量瓶中,纯化水定容后移取5ml至顶空进样瓶中,上机待测;注:考虑到异丙硫醇会有残留,进样顺序依次为:空白对照—样品溶液—Std3—Std2—Std1.测试结果1.曲线建立因Std1浓度过高,响应过载故舍弃该点;后因时间关系没能再补充一校正浓度点,最后以空白(原点)、Std3、Std2建立校正曲线: http://ng1.17img.cn/bbsfiles/images/2014/12/201412291018_529779_2167114_3.bmphttp://ng1.17img.cn/bbsfiles/images/2014/12/201412291019_529780_2167114_3.bmp 2. 样品及空白的测试谱图如下:http://ng1.17img.cn/bbsfiles/images/2014/12/201412291019_529781_2167114_3.bmp3.谱库比对:峰RT=5.86min的MS图与NIST谱库对照如下:http://ng1.17img.cn/bbsfiles/images/2014/12/201412291020_529782_2167114_3.bmp经比对,RT=5.86min的峰为目标物异丙硫醇。4.结果计算:样品测的的响应值为45.19,经校正最后得样品中异丙硫醇的含量为3.35mg/kg。小结整个试验结束后有几个心得,稍作总结希望对以后有需要测试异丙硫醇的版友们有一点点帮助吧!一. 异丙硫醇的味道非常不愉快且难以散去,整个配置过程尽量在通风橱中进行;二. 就我们的测试系统而言,异丙硫醇会有所残留,测试完毕应立即对捕集肼进行清洁以免残留;对色谱柱也应做相对老化除杂处理;三. 测试时先测空白对照,其次是样品,最后进标液(先测低浓度,后测高浓度);四. 配置标液时浓度可以设置得低一点,一是因为异丙硫醇在水中的溶解性不太好,二是异丙硫醇在MS的响应很高。若时间允许,本实验应该补充更低浓度的曲线点来校正;五.猜测wax等强极性色谱柱对异丙硫醇的分析效果更好,因本实验室的wax色谱柱用于其他项目的测试,故没有做wax与DB624色谱柱的对比。后期有条件可以尝试对比一下效果。最后,非常感谢“千层峰”和“symmacros”两位老师的鼓励和帮助!
请教各位老师:在用VGA测量汞时,前处理中残留的硝酸在小于多少含量时,才不会对测量汞产生影响?谢谢。
求助GB/T 22312-2008 塑料 聚丙烯酰胺 残留丙烯酰胺含量测定方法[em0910]
我做顶空有机残留用的是外标法,根据计算公式一直都认为求得的数据是含量,可是一个同行(比较有经验一点)说求得的是纯度不能和含量混为一谈,我晕了,想和各位色友讨论一下。







 我要推广仪器
我要推广仪器
 下载APP
下载APP