RT!哪位大侠可以分享一下“不同化合物基团在近红外区的吸收谱带”,万分感激!
各位大侠,请问红外谱图中1403处是什么基团的吸收峰,谢谢了。谱图见附件。
急!急!红外吸收光谱 1100处有个尖锐的强吸收峰可能是什么基团呢据说是一氟碳有机化合物 做了个红外 如图 偶是菜鸟 不能准确判断各吸收峰基团 请各位知晓的不吝赐教~
以前在论坛上看到过一个网站,输入红外吸收峰位置,就可以找出所有在该位置出峰的基团,后来重装电脑,找不到那个网站了,谁有好的查红外谱图的网站啊,谢谢了!
红外光谱与紫外光谱吸收峰不同希望说明为什么
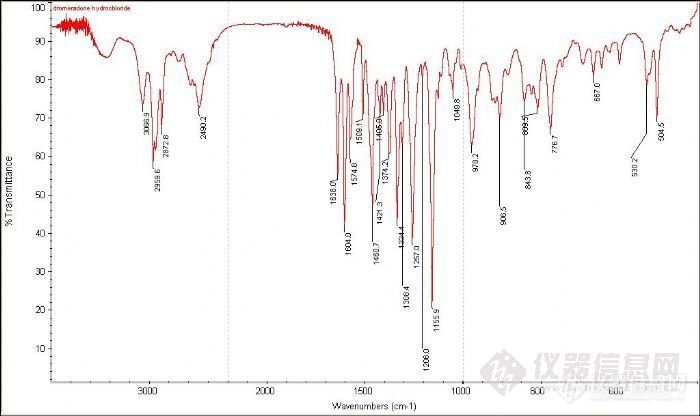
谁能帮我分析一下2490处是什么基团的吸收峰啊http://ng1.17img.cn/bbsfiles/images/2012/10/201210300846_400161_1746325_3.jpg
测一种蓝绿色溶液(应该是纯的),分别在670nm和630nm有吸收峰,630nm有最大吸收峰,请问可以断定溶液中可能含有什么基团,或者是有什么资料可以查得该溶液中可能含有的基团吗?此外,我想知道是不是特定的最大吸收波长对应特有的生色团?望大家不吝赐教。
请问:测红外时,2380cm-1吸收峰是什么基团?谢谢.
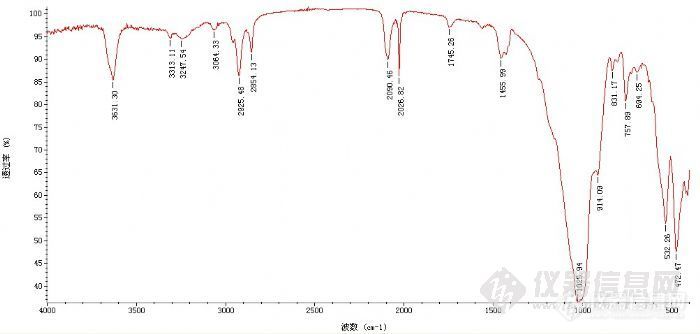
下面的谱图为一混合物谱图,主成分为无机的,想分析分析各吸收峰对应的基团,看到2000左右有点懵,求指点! 谢谢http://ng1.17img.cn/bbsfiles/images/2012/02/201202291055_351596_2315115_3.jpg
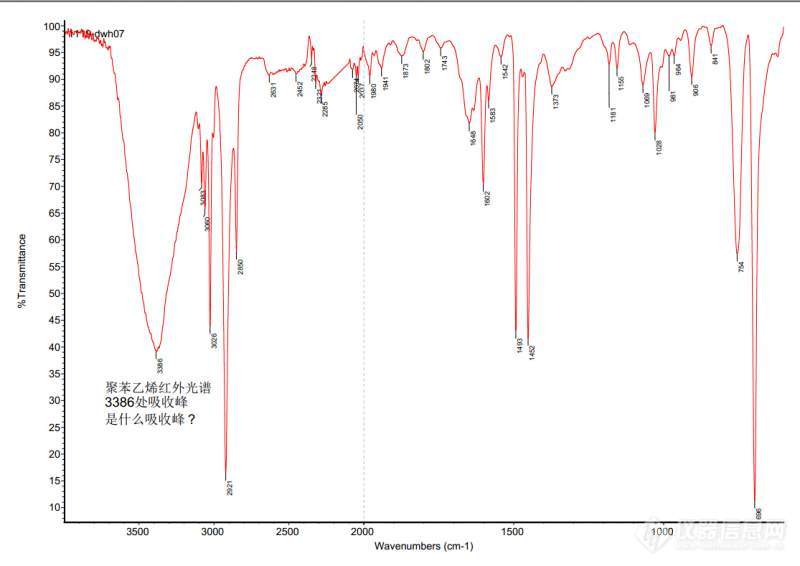
[color=#444444]求教高手,聚苯乙烯红外光谱在3386处吸收峰是什么基团啊?[/color][color=#444444][img=,690,488]https://ng1.17img.cn/bbsfiles/images/2019/09/201909271039436392_688_1646718_3.jpg!w690x488.jpg[/img][/color]
[color=#333333]同一物质不同的浓度,测紫外,最大吸收峰相同吗??[/color]
磺酸基团有紫外可见吸收吗?如果有吸收的话,磺酸羧酸形成的酸酐发生水解,这两个水解前后的磺酸基团有差异吗?非常感谢
如题:在水中对甲苯磺酸有三个吸收峰,261nm,224nm,199nm,而在PMA(丙二醇甲醚醋酸酯)中只有261的吸收峰。
[b][font=宋体]问题描述:用紫外分光光度计测叶黄素含量,国标中用无水乙醇最佳波长是[/font]445nm[font=宋体],文献中用正己烷最佳波长是[/font]474nm[font=宋体],这是因为溶剂不同造成的吗?[/font][font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])紫外光谱是用紫外光待测物紫外吸收波长与所采用的溶剂密切相关,不同溶剂对紫外吸收峰波长和强度影响不同,特别是对波长影响更大。溶剂极性对溶质最大吸收峰[/font]λ[sub]max[/sub][font=宋体]的影响与溶剂的介电常数和溶质分子的电子跃迁性质有关。[/font] [font=宋体]([/font]2[font=宋体])当用光照射时,基团中的电子吸收光能发生能级改变,形成特征的强吸收带,这些基团称作发色团,它们都含有不饱和键或未共用电子对,能产生[/font]π-π*[font=宋体]及[/font]n-π*[font=宋体]的跃迁。溶剂极性越强,由[/font]π-π*[font=宋体]跃迁产生的谱带向长波方向移动越显著。这是因为发生[/font]π-π*[font=宋体]跃迁的分子激发态的极性总是大于基态,在极性溶剂作用下,激发态能量降低的程度大于基态,从而实现基态到激发态跃迁所需能量变小,致使吸收带发生红移;所用溶剂极性越强,由[/font]n-π*[font=宋体]跃迁产生的谱带向短波方向移动越明显。这是因为发生[/font]n-π*[font=宋体]跃迁的分子都含有未成键[/font]n[font=宋体]电子,这些电子会与极性溶剂形成氢键,其作用强度是极性较强的基态大于极性较弱的激发态。因而基态能级比激发态能级的能量下降幅度大,实现[/font]n-π*[font=宋体]跃迁所需能量也响应增大,致使吸收谱带发生蓝移。[/font][font=宋体]([/font]3[font=宋体])在选择测定吸收光谱曲线的溶剂时应注意如下几点:[/font]a[font=宋体].[/font][font=宋体]能很好地溶解被测物,并形成良好化学和光化学稳定性的溶剂;[/font][font=宋体]b.[/font][font=宋体]在溶解度允许范围内,尽量选择极性较小的溶剂;[/font][font=宋体]c.[/font][font=宋体]溶剂在样品的吸收光谱区无明显吸收。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font][color=red] [/color]
同一溶剂下,带有不同取代基的化合物的紫外吸收峰发生红移的原因有哪些?
请问红外1726的波段属于什么基团吸收?样品是混合物,不确定呢!我没图,但急着要.小女子提前谢谢大虾们啦!!!
朋友们,我最近在做一批样品,有可能是含有砜基团的苯环系列。请问谁有砜的吸收谱图,我的谱图库不全。My Eail kingpeng621@sohu.com.[em61] [em61]
大家好,小女子做了些有关根系分泌物里的一些氨基酸类物质的分析。一开始自己摸索方法用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]做,但是因为水平有限,没做出来。后来有一天用HPLC试了一下结果出现了一个峰(纯化后的样品)且只有一个峰,用标准样品试了后也是只有这一个峰。不知道这样算不算是,而且我是用的紫外检测器测出来的。由于以前用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]一直没有做出来,现在用液相一下就出来了,我心里很毛,不敢确定是不是,所以请教大家一下什么基团有紫外吸收特性,有这方面的资料或书吗,请推荐一下?我的样品是个CN杂环,有氨基,羧基,羟基。谢谢了。
我是拿一个相同的物质在提纯后于不同的时间测定紫外吸收,但是在不同的时间内分别出现为:1、228,2742、297第二种情况很奇怪,连B带也没有了,为什么啊?非常紧急,希望及时得到答复,谢谢!
同一种梯度洗脱,出峰时间和标品相同,但最大吸收波长不同,能不能判断为同一物质?,谢谢指导啊
请问各位大侠,含有酰胺基团的二肽有没有紫外吸收呢?
跟大家分享一下,聚氨酯的IR分析 v3250-3500 ms OH伸缩振动、NHCO的顺式NH伸缩振动。 v2940、2860 s CH2、CH3伸缩振动。 v2240-2280 s NCO特征吸收峰。 v2120 s 碳化二亚胺吸收峰。 v1770-1785 s 脲二酮环(二聚体)中的C=O。 v1715-1750 vs 酯基C=O、酰胺I键C=O。 v1689-1710 s 异腈脲酸酯(三聚体)中C=O(1408-1430也有峰) v1600-1615 苯环C=C骨架伸缩振动。 v1520-1560 ms 酰胺II键(N-H)变形振动。 v1450-1470 CH2变形振动、CH3非对称变形振动。 v1380 CH3对称变形振动 v1225-1235 聚酯C-O伸缩或OH变形振动 v1060-1150 宽s C-O-C(脂肪族醚)吸收峰。
大家好,小女子做了些有关根系分泌物里的一些氨基酸类物质的分析。一开始自己摸索方法用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]做,但是因为水平有限,没做出来。后来有一天用HPLC试了一下结果出现了一个峰(纯化后的样品)且只有一个峰,用标准样品试了后也是只有这一个峰。不知道这样算不算是,而且我是用的紫外检测器测出来的。由于以前用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]一直没有做出来,现在用液相一下就出来了,我心里很毛,不敢确定是不是,所以请教大家一下什么基团有紫外吸收特性,有这方面的资料或书吗,请推荐一下?我的样品是个CN杂环,有氨基,羧基,羟基。谢谢了。
有什么基团或者物质在700-800nm左右有特征吸收光谱呢?常见的光谱分析讲的都是300nm左右。谢谢各位大侠。
紫外光谱吸收带的分类总觉得这一块被忽略了,所以赶紧弄上来,唤起大家的回忆.原来感觉四谱分析(红外、紫外、质谱和核磁)在有机分析中一直占据着主导地位,但现在感觉紫外光谱一直被人们所忽视,一直没想明白怎么回事。前一段时间参加仪器展览的时候,听一老师讲多极质谱,原来可以代替四谱分析来解决问题。突然明白怎么回事,也感觉自己已经赶不上时代了,知识的更新速度远比俺学习的速度快的多。感慨之余,和大家一起来学习分享紫外光谱吸收带的一些问题。紫外及可见光谱包括有几个谱带系,不同的谱带系相当于不同电子能级的跃迁。俺以前结构化学没有学好,现在很后悔啊!!!1、远紫外(真空紫外)吸收带这一块用的比较少,应该是非常少,一般紫外分光光度计的波长都是从200纳米开始的,因为远紫外(真空紫外)吸收带被空气强烈吸收,顾名思义,也叫真空紫外。主要是烷烃化合物的吸收带,如C-C、C-H基团中,为δ→δ*跃迁,最大吸收波长小于200纳米,范围在10-200纳米。2、尾端吸收带饱和卤代烃、胺类或含杂原子的单键化合物的吸收带,由于这类化合物含有一个或几个孤对电子,因此产生n→δ*跃迁,其范围从远紫外区末端到近紫外区,在200纳米附近。所以,一般在紫外区扫描或全波长扫描的时候,建议从210纳米开始,因为很多物质都存在末端吸收,多扫了没有多大意义,从节省时间和氘灯的角度考虑,建议从210纳米开始扫描。3、R带这个吸收属于弱吸收带,但是溶剂效应比较明显,所以俺在此友情提醒,在选择溶剂的时候一定要注意哦。R带是共轭分子的含杂原子基团的吸收带,如C=O,N=O,N=N等基团,有n→π*跃迁产生,为弱吸收带,摩尔吸光系数K一般小于100L.mol-1.cm-1;随着溶剂极性的增加,R带会发生蓝移,附近如有强吸收带,R带有时会红移,有时可能观察不到。4、K带这个用的比较多,也是有机物定性定量的基础,其最大吸收往往是由K带决定的,一般来说,如果某物质存在共轭双键,从理论上来将都可以用紫外去定性定量的,所以俺建议大家,要特别注意K带呀。共轭体系的π→π*跃迁所产生的吸收带,如共轭烯烃,烯酮等。K带的吸收强度很高,一般K大于10000L.mol-1.cm-1。5、B带理论支持:芳香和杂环化合物π→π*的特征吸收带。苯的B吸收带在230-270纳米之间,并出现包含有多重峰或精细结构的宽吸收带(这也是为什么有馒头峰的原因)。但取代芳香烃的B带精细结构会消失,极性溶剂也会使精细结构消失。6、E带含有苯环的物质一般在B带有和E带吸收,但是俺做过试验,感觉B带的吸收远远K带强烈,就以山梨酸和苯甲酸为例,相同浓度的山梨酸的吸收特别强烈,最大吸收很明显,可是苯甲酸的却象馒头峰,最大吸收特不明显,只有通过求导才能找出最大吸收来,比较郁闷。这也可以从吸光系数看出来,B带的吸光系数为250-300 L.mol-1.cm-1,感觉不是很灵敏。E带吸收系数大,但由于E和B的作用,往往峰形不太好,不利于分析。也属于芳香结构的特征吸收,由处于环状共轭的三个乙烯键的苯型体系中的π→π*跃迁所产生。E带又分为E1和E2带。E带属于强吸收带,K大于10000 L.mol-1.cm-1

吸收窗、峰宽、阈值的应用实例摘要:吸收窗、峰宽、阈值等用来对色谱峰进行判定或积分,这些专用词语对于初次接触气相的人员来说,太过抽象难懂,不清楚这些值设定多大为最佳,也不清楚它的实际应用,下面讲几个应用实例。1 实验部分1.1 仪器与试剂瓦里安气相GC450,PFPD、ECD检测器,自动进样器,100位进样盘。多用于蔬菜中农药残留,有机磷与有机氯的检测。DB-5(30*0.25*0.25)柱检测有机磷,VF-1(30*0.32*0.25)柱检测有机氯。1.2实验方法一、吸收窗:瓦里安气相软件上的吸收窗,是指保留时间,保留时间是从进样开始至每个组分流出曲线达极大值(峰顶)所需时间,可作为色谱峰位置的标志。软件上默认的为吸收窗为0.20min,也就是说±0.20min以内都可默认为该物质。进四种有机磷标液:乐果、二唪农、马拉硫磷、甲基异硫磷等,这张图谱设为模板,模板中的吸收窗设定为0.20min。http://ng1.17img.cn/bbsfiles/images/2014/10/201410031801_516849_1645480_3.jpg吸收窗设为0.20min。http://ng1.17img.cn/bbsfiles/images/2014/10/201410031801_516850_1645480_3.jpg这张标液模板的结果。http://ng1.17img.cn/bbsfiles/images/2014/10/201410031802_516851_1645480_3.jpg1号样品出的图谱,以标液模板为对照,自动计算,得出的结果。http://ng1.17img.cn/bbsfiles/images/2014/10/201410031802_516852_1645480_3.jpg这时将标液中乐果的吸收窗改为0.05。http://ng1.17img.cn/bbsfiles/images/2014/10/201410031803_516853_1645480_3.jpg以修改过吸收窗的模板去计算同一个样品,可以看出乐果变为未知峰。从上面这个例子可以看出,当样品中的出峰保留时间与标液出峰时间不相符时,吸收窗设定的范围不同,定性的结果也不同,乐果的出峰时间差异为0.06min,当吸收窗设定为0.2min时,可以判定为乐果,当吸收窗设定为0.05min时,样品中乐果就不存在了。那么实际工作中将吸收窗设定为多少合适呢?工作中每天气体流速不同,温度与湿度不同,仪器状况也稍有差异,所以保留时间有一定的漂移是正常的,一般会设定为0.2min,如果判定为该物质,还需要用双柱定性或加标来判断,最好有气质来定性,准确度会更高。如果有大批量样品处理,例如近百个样品,第一个样品与第一百个样品的出峰时间会有差异,这时会选择每进20个样品进一次标液,用标液图去对照这20个样品,看是否有检出,进一个标液去对照近百个样品,这样的做法会有较大误差。
红外光谱基团频率分析及应用 基团频率和特征吸收峰物质的红外光谱是其分子结构的反映,谱图中的吸收峰与分子中各基团的振动形式相对应。多原子分子的红外光谱与其结构的关系,一般是通过实验手段得到。这就是通过比较大量已知化合物的红外光谱,从中总结出各种基团的吸收规律。 实验表明,组成分子的各种基团,如O-H、N-H、C-H、C=C、C=OH和C C等,都有自己的特定的红外吸收区域,分子的其它部分对其吸收位置影响较小。通常把这种能代表及存在、并有较高强度的吸收谱带称为基团频率,其所在的位置一般又称为特征吸收峰。一、基团频率区和指纹区(一)基团频率区 中红外光谱区可分成4000 cm-1 ~1300 cm-1和1800cm-1 (1300 cm-1 )~ 600 cm-1两个区域。最有分析价值的基团频率在4000 cm-1 ~ 1300 cm-1 之间,这一区域称为基团频率区、官能团区或特征区。区内的峰是由伸缩振动产生的吸收带,比较稀疏,容易辨认,常用于鉴定官能团。 在1800 cm-1 (1300 cm-1 )~600 cm-1 区域内,除单键的伸缩振动外,还有因变形振动产生的谱带。这种振动与整个分子的结构有关。当分子结构稍有不同时,该区的吸收就有细微的差异,并显示出分子特征。这种情况就像人的指纹一样,因此称为指纹区。指纹区对于指认结构类似的化合物很有帮助,而且可以作为化合物存在某种基团的旁证。基团频率区可分为三个区域:LT7U 键或芳香核共轭时,该峰位移到2220~2230 cm-1附近。若分子中含有C、H、N原子, -C N基吸收比较强而尖锐。若分子中含有O原子,且O原子离-C N基越近, -C N基的吸收越弱,甚至观察不到。1900~1200 cm-1为双键伸缩振动区 该区域重要包括三种伸缩振动: ① C=O伸缩振动出现在1900~1650 cm-1 ,是红外光谱中很特征的且往往是最强的吸收,以此很容易判断酮类、 醛类、酸类、酯类以及酸酐等有机化合物。酸酐的羰基吸收带由于振动耦合而呈现双峰。② C=C伸缩振动。烯烃 的C=C伸缩振动出现在1680~1620 cm-1 ,一般很弱。单核芳烃的C=C伸缩振动出现在1600 cm-1和1500 cm-1附近,有两个峰,这是芳环的骨架结构,用于确认有无芳核的存在。③ 苯的衍生物的泛频谱带,出现在2000~1650 cm-1范围, 是C-H面外和C=C面内变形振动的泛频吸收,虽然强 度很弱,但它们的吸收面貌在表征芳核取代类型上是有用的。(二)指纹区d 1. 1800(1300)~900 cm-1区域是C-O、C-N、C-F、C-P、C-S、 P-O、Si-O等单键的伸缩振动和C=S、S=O、P=O等双键的伸缩振动吸收。 其中 1375 cm-1的谱带为甲基的 C-H对称弯曲振动,对识别甲基十分有用,C-O的伸缩振动在1300~1000 cm-1 ,是该区域最强的峰,也较易识别。 900~650 cm-1区域的某些吸收峰可用来确认化合物的顺反构型。 例如,烯烃的=C-H面外变形振动出现的位置,很大程度上决定于双键的取代情况。对于RCH=CH2结构,在990 cm-1和910 cm-1出现两个强峰;为RC=CRH结构是,其顺、反构型分别在690 cm-1和970 cm-1出现吸收峰,可以共同配合确定苯环的取代类型。二、常见官能团的特征吸收频率三、影响基团频率的因素 基团频率主要是由基团中原子的质量和原子间的化学键力常数决定。然而,分子内部结构和外部环境的改变对它都有影响,因而同样的基团在不同的分子和不同的外界环境中,基团频率可能会有一个较大的范围。因此了解影响基团频率的因素,对解析红外光谱和推断分子%( 结构都十分有用。 影响基团频率位移的因素大致可分为内部因素和外部因素。 内部因素:1. 电子效应 包括诱导效应、共轭效应和中介效应,它们都是由于化学键的电子分布不均匀引起的。(1)诱导效应(I 效应) 由于取代基具有不同的电负性,通过静电诱导作用,引起分子中电子分布的变化。从而改变了键力常数,使基团的特征频率发生了位移。 例如,一般电负性大的基团或原子吸电子能力强,与烷基酮羰基上的碳原子数相连时,由于诱导效应就会发生电子云由氧原子转向双键的中间,增加了C=O键的力常数,使C=O的振动频率升高,吸收峰向高波数移动。随着取代原子电负性的增大或取代数目的增加,诱导效应越强,吸收峰向高波数移动的程度越显著。(2)中介效应(M效应)当含有孤对电子的原子(O、S、N等)与具有多重键的原子相连时,也可起类似的共轭作用,称为中介效应。由于含有孤对电子的原子的共轭作用,使C=O上的电子云更移向氧原子,C=O双键的电子云密度平均化,造成C=O键的力常数下降,使吸收频率向低波数位移。 对同一基团,若诱导效应和中介效应同时存在,则振动频率最后位移的方向和程度,取决于这两种效应的结果。当诱导效应大于中介效应时,振动频率向高波数移动,反之,振动频率向低波数移动。 2 . 氢键的影响氢键的形成使电子云密度平均化,从而使伸缩振动频率降低。游离羧酸的C=O键频率出现在1760 cm-1 左右,在固体或液体中,由于羧酸形成二聚体, C=O键频率出现在1700 cm-1 。 分子内氢键不受浓度影响,分子间氢键受浓度影响较大。 3. 振动耦合 当两个振动频率相同或相近的基团相邻具有一公共原子时,由于一个键的振动通过公共原子使另一个键的长度发生改变,产生一个“微扰”,从而形成了强烈的振动! 相互作用。其结果是使振动频率发生感变化,一个向高频移动,另一个向低频移动,谱带分裂。振动耦合常出现在一些二羰基化合物中,如,羧酸酐。4.Fermi共振 当一振动的倍频与另一振动的基频接近时,由于发生相互作用而产生很强的吸收峰或发生裂分,这种现象称为Fermi共振。外部因素 外部因素主要指测定时物质的状态以及溶剂效应等因素。 同一物质的不同状态,由于分子间相互作用力不同,所得到光谱往往不同。 分子在气态时,其相互作用力很弱,此时可以观察到伴随振动光谱的转动精细结构。 液态和固态分子间作用力较强,在有极性基团存在时,可能发生分子间的缔合或形成氢键,导致特征吸收带频率、强度和形状有较大的改变。例如,丙酮在气态时的 C-H为1742 cm-1 ,而在液态时为1718 cm-1 。 在溶液中测定光谱时,由于溶剂的种类、溶剂的浓度和测定时的温度不同,同一种物质所测得的光谱也不同。通常在极性溶剂中,溶质分子的极性基团的伸缩振动频率随溶剂极性的增加而向低波数方向移动,并且强度增大。因此,在红外光谱测定中,应尽量采用非极性的溶剂。
一个综合的FT-IR资源,有吸收峰位搜索窗口,有可下载的资源等....http://infrared.als.lbl.gov/FTIRinfo.html连接上面的网页,如果您想搜索1540吸收峰可能是什么基团的吸收,可以将1540输入下面IR Wizzard: Enter a peak's的方框中,点击submit.IR Wizzard: Enter a peak's location in cm-1: 就会出现40个结构片段和吸收峰的位置,还有强度注明,选中你要的1515 up to 1570(s Amide II ) ,击click here,会有更进一步的信息供你参考.
物质吸收的光源不同出现的色散谱面也不同 物质吸收的光源不同,出现的色散谱面也不同。这是1752年苏格兰人梅耳维尔在实验中发现的,因梅耳维尔死得过早(20多岁),后人沒做进一步的研究,只注意谱线及应用上去了,目前我也没看到或搜索到这方面的讨论文。 光源不同色散潽面也不同:当两个不相同的物质(一个是白色,另一个是黑色)拼在一起时,若在热辐射光源下(炽热的固体.液体发光),则在两物质相交处出现的色散象是连续(各波长)无界段的谱面,也就是现在的连续光谱象。若如这两个不同物质在充有稀薄气体(如荧光光源.高压气体发光),则出现的色散谱面(各单色)是一段一段的,各单色之间有明显的过渡界。 物质在不同光源下出现的两种不同色散谱面是一个末解决的问题,望各位去做一下这个实验,也许这里埋藏着一些重要的发现。
紫外光谱吸收带的分类总觉得这一块被忽略了,所以赶紧弄上来,唤起大家的回忆.原来感觉四谱分析(红外、紫外、质谱和核磁)在有机分析中一直占据着主导地位,但现在感觉紫外光谱一直被人们所忽视,一直没想明白怎么回事。 前一段时间参加仪器展览的时候,听一老师讲多极质谱,原来可以代替四谱分析来解决问题。突然明白怎么回事,也感觉自己已经赶不上时代了,知识的更新速度远比俺学习的速度快的多。感慨之余,和大家一起来学习分享紫外光谱吸收带的一些问题。紫外及可见光谱包括有几个谱带系,不同的谱带系相当于不同电子能级的跃迁。俺以前结构化学没有学好,现在很后悔啊!!!1、远紫外(真空紫外)吸收带 这一块用的比较少,应该是非常少,一般紫外分光光度计的波长都是从200纳米开始的,因为远紫外(真空紫外)吸收带被空气强烈吸收,顾名思义,也叫真空紫外。主要是烷烃化合物的吸收带,如C-C、C-H基团中,为δ→δ*跃迁,最大吸收波长小于200纳米,范围在10-200纳米。2、尾端吸收带 饱和卤代烃、胺类或含杂原子的单键化合物的吸收带,由于这类化合物含有一个或几个孤对电子,因此产生n→δ*跃迁,其范围从远紫外区末端到近紫外区,在200纳米附近。 所以,一般在紫外区扫描或全波长扫描的时候,建议从210纳米开始,因为很多物质都存在末端吸收,多扫了没有多大意义,从节省时间和氘灯的角度考虑,建议从210纳米开始扫描。3、R带 这个吸收属于弱吸收带,但是溶剂效应比较明显,所以俺在此友情提醒,在选择溶剂的时候一定要注意哦。 R带是共轭分子的含杂原子基团的吸收带,如C=O,N=O,N=N等基团,有n→π*跃迁产生,为弱吸收带,摩尔吸光系数K一般小于100L.mol-1.cm-1;随着溶剂极性的增加,R带会发生蓝移,附近如有强吸收带,R带有时会红移,有时可能观察不到。4、K带 这个用的比较多,也是有机物定性定量的基础,其最大吸收往往是由K带决定的,一般来说,如果某物质存在共轭双键,从理论上来将都可以用紫外去定性定量的,所以俺建议大家,要特别注意K带呀。共轭体系的π→π*跃迁所产生的吸收带,如共轭烯烃,烯酮等。K带的吸收强度很高,一般K大于10000L.mol-1.cm-1。5、B带 理论支持:芳香和杂环化合物π→π*的特征吸收带。苯的B吸收带在230-270纳米之间,并出现包含有多重峰或精细结构的宽吸收带(这也是为什么有馒头峰的原因)。但取代芳香烃的B带精细结构会消失,极性溶剂也会使精细结构消失。6、E带 含有苯环的物质一般在B带有和E带吸收,但是俺做过试验,感觉B带的吸收远远K带强烈,就以山梨酸和苯甲酸为例,相同浓度的山梨酸的吸收特别强烈,最大吸收很明显,可是苯甲酸的却象馒头峰,最大吸收特不明显,只有通过求导才能找出最大吸收来,比较郁闷。这也可以从吸光系数看出来,B带的吸光系数为250-300 L.mol-1.cm-1,感觉不是很灵敏。E带吸收系数大,但由于E和B的作用,往往峰形不太好,不利于分析。 也属于芳香结构的特征吸收,由处于环状共轭的三个乙烯键的苯型体系中的π→π*跃迁所产生。E带又分为E1和E2带。E带属于强吸收带,K大于10000 L.mol-1.cm-1 [img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=71804]吸收带理论[/url]







 我要推广仪器
我要推广仪器
 下载APP
下载APP