高分子表征技术专题——小角中子散射技术及其在大分子结构表征中的应用
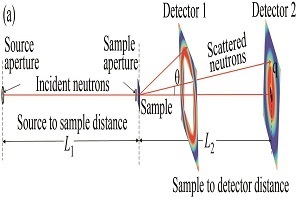
2021年,《高分子学报》邀请了国内擅长各种现代表征方法的一流高分子学者领衔撰写从基本原理出发的高分子现代表征方法综述并上线了虚拟专辑。仪器信息网在获《高分子学报》副主编胡文兵老师授权后,也将上线同名专题并转载专题文章,帮助广大研究生和年轻学者了解、学习并提升高分子表征技术。在此,向胡文兵老师和组织及参与撰写的各位专家学者表示感谢。更多专题内容详见:高分子表征技术专题高分子表征技术专题前言孔子曰:“工欲善其事,必先利其器”。 我们要做好高分子的科学研究工作,掌握基本的表征方法必不可少。每一位学者在自己的学术成长历程中,都或多或少地有幸获得过学术界前辈在实验表征方法方面的宝贵指导!随着科学技术的高速发展,传统的高分子实验表征方法及其应用也取得了长足的进步。目前,中国的高分子学术论文数已经位居世界领先地位,但国内关于高分子现代表征方法方面的系统知识介绍较为缺乏。为此,《高分子学报》主编张希教授委托副主编王笃金研究员和胡文兵教授,组织系列从基本原理出发的高分子现代表征方法综述,邀请国内擅长各种现代表征方法的一流高分子学者领衔撰写。每篇综述涵盖基本原理、实验技巧和典型应用三个方面,旨在给广大研究生和年轻学者提供做好高分子表征工作所必须掌握的基础知识训练。我们的邀请获得了本领域专家学者的热情反馈和大力支持,借此机会特表感谢!从2021年第3期开始,以上文章将陆续在《高分子学报》发表,并在网站上发布虚拟专辑,以方便大家浏览阅读. 期待这一系列的现代表征方法综述能成为高分子科学知识大厦的奠基石,支撑年轻高分子学者的茁壮成长!也期待未来有更多的学术界同行一起加入到这一工作中来.高分子表征技术的发展推动了我国高分子学科的持续进步,为提升我国高分子研究的国际地位作出了贡献. 借此虚拟专辑出版之际,让我们表达对高分子物理和表征学界的老一辈科学家的崇高敬意!小角中子散射技术及其在大分子结构表征中的应用The Basic Principle of Small Angle Neutron Scattering and Its Application in Macromolecules作者:左太森,马长利,韩泽华,李雨晴,李明涛,程贺作者机构:中国科学院高能物理研究所 中国散裂中子源 2.散裂中子源科学中心,东莞,523803 中国科学院大学,北京,100049作者简介:程贺,男,1978年生. 中国科学院高能物理研究所东莞研究部研究员. 1996年考取中国科学技术大学,2006年在吴奇教授课题组获得博士学位. 随后赴中国科学院化学研究所韩志超研究员课题组工作,建设我国第一台SANS(2012年国家验收). 2014年加入中国散裂中子源,中国科学院高能物理研究所东莞研究部,现正在主持建设世界上第二台基于散裂中子源的VSANS. 致力于使用和发展散射方法,研究软物质多相多尺度结构和动态学行为.摘要小角中子散射(SANS)是一种表征从纳米到微米尺寸物质特征结构的有力工具,配合中子的强穿透性和同位素辨识等特性,在软物质大分子结构表征方面发挥着独特的作用. 随着中国散裂中子源(CSNS)在2018年正式对外接受机时申请,国内SANS用户群逐年扩大. 本文首先简要介绍小角中子散射技术的基本原理、谱仪结构和实验技巧,然后紧扣小角谱仪的特点和方法学方面的最新进展,介绍小角中子散射在高分子溶液、高分子共混物和复合材料、高分子结晶、凝胶、多孔材料、生物大分子等研究领域的结构表征方面的典型应用. 小角中子散射和其他表征手段,如小角X射线散射(SAXS)相互紧密配合和补充,成为连接大分子内部多相多尺度的微观结构和宏观性的桥梁.AbstractSmall angle neutron scattering (SANS) is a powerful tool to characterize multi-scale structures in macromolecules. Deep penetration and H/D isotope labeling make it a unique scattering method. To make it more familiar to the users, basic principle of SANS, instrumentation and experimental skills were firstly demonstrated. Then typical applications in the fields of polymer solution, polymer blends, nanocomposites, crystallization, gels, porous materials and biomacromolecules were introduced. As for the data analysis of complex systems, such as biomacromolecules, in addition to the traditional data analysis methods, advanced methods such as the ab initial analysis and Reverse Monte-Carlo (RMC) simulations provide more detailed information. Combine with small angle X-ray scattering (SAXS), static light scattering (SLS), electron microscope (EM)et al., SANS enables us to solve the structure and interaction of more complicated systems such as interaction of biomacromolecues and solvation of polymers in mixed solutions. As the China Spallation Neutron Source (CSNS) was officially opened to the users around the world in 2018 and SANS instruments equipped with various sample environments are being built, more opportunities are opened to the SANS communities domestically and abroad.关键词小角中子散射 大分子 多相多尺度 结构表征 中国散裂中子源KeywordsSmall angle neutron scattering Macromolecules Multi-scale and multi-phase Structure characterization China spallation neutron source 小角散射,通常包括小角光散射(SLS)、小角X射线散射(SAXS)和小角中子散射(SANS),都是表征物质纳米到微米的多尺度特征结构的有力手段[1,2]. 它们的基本原理[3]和数据处理分析方法[4]十分类似,三者可以互补和互相验证. 3种散射方法有两点主要不同之处:一是光源与样品的作用机理不同,所以使用不同散射方法时样品的衬度不同;二是波长不同,所以研究的特征尺度范围不同. 首先,衬度直接决定了散射实验的可行性. 光散射衬度来自样品的微分折光指数;X射线与核外电子相互作用,衬度来自于电子云密度,所以原子序数高的元素衬度高;对于中子,由于中子直接作用于原子核,与核的性质有关而与原子序数无关,反而同一元素的各种同位素的中子衬度有很大不同. 小角中子散射的衬度等于样品与分散剂的相干散射长度密度之差,这里的相干散射长度密度(ρcoh,单位:Å-2)是散射体中所有的元素或同位素的相干散射长度(bcoh, 单位:Fermi,1 Fermi = 10-15 m)的加权平均与散射体的摩尔体积之比;同位素的散射截面相当于原子核与中子相互作用被散射的概率( σσ,单位barn, 1 barn = 10-24 cm 2),正比于散射长度的平方. 中子与原子核相互作用,除了被散射外,还会有一定的概率被吸收. 常见天然元素和同位素对于1.8 Å中子的相干散射长度、相干和非相干散射截面以及吸收截面的数据如表1所示[5]. 设计SANS实验的第一步需要估算样品的中子衬度和透光率,前者决定了SANS实验的可行性,后者决定了数据分析的可行性. 根据表1,已知大分子体系的元素、同位素组成和密度,可以计算中子衬度,溶液体系衬度为溶质和溶剂的中子相干散射长度密度差,二元共混体系衬度为二元组分大分子的中子相干散射长度密度差. 衬度低的样品无法进行SANS实验(比如一般的非晶碳氢化合物样品,化学组成一般为CH2,根据表1,bc+2bH≈0bc+2bH≈0,在不进行氘代的情况下无法进行SANS实验);而样品对中子的透过率可以通过式(1)所示的朗伯-比尔定律计算.其中:d为样品厚度.nini为样品中第ii种元素的原子比例,pij、σij(λ)σij(λ)和ρijρij分别为第i种元素的第j种同位素的丰度、全截面和数密度. 其中全截面包含相干、非相干和吸收截面,同位素截面相关数据可以参考ENDF数据库[6]. 传统的散射基本理论是建立在单次散射的基础上的,如果样品太厚,透光率较低,可能在实验中引入多次散射,造成数据无法用常规分析方法解析,所以一般的SANS实验要求 Ttrans85%,如果是溶液样品,尽量采用氘代溶剂.Table 1Coherent scattering length and coherent, incoherent and adsorption scattering cross section of common elements in macromolecules and commonly used isotopes in SANS experiments[5].一些吸收截面非常大的天然元素或者同位素通常用于中子吸收材料,如表1中的B-10,在实验样品中要尽量避免这类对热中子具有强吸收的同位素,除B-10外,还有Cd-113、Gd-155、Gd-157、Sm-149、Eu-151等同位素.对于结构表征的各类技术,能够覆盖的尺寸范围很大程度上决定了这一技术的应用范围. 用于光散射的激光波长在可见光范围,所以小角激光光散射观察尺度在微米的数量级,而静态激光散射的观察尺度在20~300 nm;由于X射线和中子的波长在埃的数量级,所以常规的SAXS和SANS可以测量1~300 nm的特征尺度.表2总结了3种小角散射方法的一些基本特征,可以看到每种方法都有其特点和不足. 小角光散射波长较长,需要样品透明并且容易受到灰尘的影响;小角X射线散射的优势是亮度非常高,特别是同步辐射X射线小角,缺点是穿透能力一般,容易被吸收(当然共振散射赋予了它另外的特点);小角中子散射的特点是穿透能力强,可以加载各类样品环境,同时还能够识别同位素,可以得到样品的绝对散射强度,缺点是中子源亮度太低. 所以实际使用中,用户需要依据自身样品的特点和需要观察的特征尺度范围,选择合适的散射手段,互相验证和补充.Table 2Comparison between SLS, SAXS and SANS.随着小角中子散射方法的应用越来越广泛,谱仪和方法学上出现了2种趋势,一方面通过中子束的聚焦或准直向更小散射矢量方向扩展1~2个量级,研究特征尺度更大的体系,典型的就是发展微小角(VSANS)[7]甚至超小角(USANS)中子散射谱仪[8];另一方面利用波长更短的中子的散射将散射矢量扩展到50 Å-1以上,研究无序体系在原子尺度上的结构,即所谓的无序大分子中子全散射方法[9]. 谱仪技术发展的驱动力在于实现通过一次散射实验来表征样品从原子到分子,再到组装体,甚至相区的多相多尺度结构的梦想. 虽然这些谱仪的设计思路和物理结构千差万别,但是它们的基本散射原理完全相同. 下文将着重介绍SANS谱仪.1小角中子散射谱仪、基本原理、实验技术和方法小角中子散射谱仪通常分为两类,一类是基于反应堆的固定波长小角谱仪[10],国内有绵阳研究堆的狻猊谱仪和中国先进研究堆的小角中子散射谱仪;另一类是基于强流脉冲中子源的飞行时间小角谱仪[11],国内有CSNS的小角中子散射谱仪. 固定波长小角谱仪,利用速度选择器将中子单色化后进行散射实验;而飞行时间小角谱仪则采用白光中子进行散射实验,利用脉冲中子从中子源运动到探测器的飞行时间标定中子波长. 两类SANS的基本原理完全一样,准直系统通常为如图1所示的小孔几何,源光阑和样品光阑用于中子准直,1个或者多个探测器接收散射中子[7].Fig. 1(a) Schematic diagram of the SANS instrument (b) The relationship between the characteristic length scaled and the scattering vector q⇀q⃑ (Bragg's Law). 运动的中子从量子力学的观点可以看成一种物质波,其波长λ = h/(mnv)(其中h为普朗克常数,mn为中子质量,v为中子速度),入射中子的波矢量记作k⇀i,其绝对值为2π/λ,中子被样品散射后,散射波矢量记作k⇀s,如果是弹性散射,中子波长不变,其绝对值仍为2π/λ.散射前后,入射波矢量和散射波矢量的差值k⇀s−k⇀i定义为散射矢量q⇀.图1是CSNS的VSANS谱仪在小角模式下的示意简图. 根据如图 1所示的几何关系和矢量加减规则得到布拉格公式:其中θ为散射角. 如果样品的特征长度为d,根据如图1几何关系和布拉格方程,两束被样品散射的中子的波程差为2dsin(θ/2),当波程差等于波长λ的整数倍时,散射中子相干增强,即:当n取1时,由公式(4)可知,正空间的样品特征长度与散射矢量q是倒易关系,即1/q是正空间的尺子,在计划实验时,需要对样品的特征尺寸范围有一个预判. 根据香农采样定理[12]:如果谱仪q范围为0.001~0.3 Å-1,其可表征的样品特征尺寸范围为300~1 nm. 如果能将中子聚焦,或者放弃一个方向的分辨率,将最小q向低q方向推进1~2个量级,从而能够表征的样品的特征尺度将增加1~2个量级. 我们将这类谱仪称为微小角中子散射谱仪(qmin=10-4 Å -1)[7]和超小角中子散射谱仪(qmin=10-5 Å -1)[13].考察一个由N个大分子链组成的链间有相互作用的体系,假设每根链聚合度为n,并粗粒化单体作为基本的散射单元. 为了方便表示,如图2所示,考察体系中的链α和链β. 链α和链β的质心距离坐标原点分别为Rα和Rβ,链α和第i个单体距离链α的质心为Sαi,链β的第j个单体距离链β的质心为Sβj,链α和链β之间的距离为Rαβ,i,j距离原点分别为rαi和rβj. 根据散射基本原理,中子入射到单个单体后形成球面波,其散射振幅:Fig. 2Schematic draw of the polymer chain and the vectors between atoms and polymers.一条链的散射振幅:考虑大分子与周围介质的散射长度密度差为Δρ,大分子单体的体积为υ,体系总体积为V.α和β遍历体系中的每一根链,i,j遍历链的每一个单体,得到体系的宏观散射截面可表示为公式(8).公式(8)右边第2项可以近似为倒易2根链的质心相互作用的相干散射得到公式(9).根据如图2所示的几何关系,代入(9)得到:其中F(q)为形状因子的散射振幅,定义单粒子的形状因子P(q),注意,这里的i,j位于同一个散射体或者同一条链上.散射体可近似视为连续介质,P(q)可改写为:其中,Vpart为散射体的体积,ρpart(r)为散射体内部的密度空间分布.定义散射体之间的结构因子SI(q),式(11)适用于所有散射体系对于密度分布均匀的散射体,∣∣F(q)2∣∣=|F(q)|2,而这里的dΣ(q)dΩ是散射矢量为q时的绝对散射强度(单位为cm-1). 小角中子散射实验中,经过样品散射进入立体角为ΔΩ的探测器的中子计数Is(q)(单位为count/s)与q的关系为:其中T(λ)为样品透过率,d为样品厚度,定义入射中子强度I0(λ):Φ(λ)为入射中子波长分布,ε(λ)为探测器效率,A为样品光阑面积,t为数据采集时间.所以对于典型的小角散射实验,如果实验的q值范围已经覆盖了样品的多相多尺度结构,通过一次SANS实验,可以得到Δρ(衬度),n(分子量),P(q) (基本形状)和SI(q) (相互作用),但需要注意的是SANS用了一个粗粒化的模型,所能观察的最小尺度是π/qmax,一般不小于1 nm.2小角中子散射实验一个完整的小角中子散射实验过程包括(1)计划实验:根据科学目标准备合适大小和数量的样品;(2)确定实验方案,并采集小角中子散射数据;(3)对散射数据进行处理和分析.2.1样品准备和要求在样品准备阶段需要注意几个问题,第一,衬度:样品中散射体与周围介质的散射长度密度的差异是否足够. 一般而言,如果衬度Δρ≥1×10-6 Å -2就完全没有问题,否则就需要与谱仪科学家进行沟通,依据谱仪本身的信噪比进行调整. 如果衬度不够就可能需要对溶剂或者散射体进行氘代. 第二,样品的特征尺寸是否在谱仪的测量范围内,通常谱仪的测量范围在π/qmax到π/qmin内;第三,做一些前置实验,如小角X射线散射、电镜等确定合成的样品状态是否由于聚集、结晶等过程的发生而改变. 此外,还需要注意样品的使用量和样品厚度. 根据样品内散射体的尺寸和与周围介质之间的衬度,样品量从300~1500 mg不等,样品厚度根据散射强度选择,通常为1和2 mm. 对于强散射样品,如果样品太厚会产生多重散射;对于溶液样品需要注意样品的结构与浓度有关,稀、亚浓和浓溶液结构会随着样品间相互作用而改变,为区分P(q)和SI(q)对I(q)的影响,除硬球体系之外,一般需要在稀溶液中先确定样品P(q),这时也许需要在0.1 wt%~5 wt%之间做多个样品,从而外推到无限稀溶液的情况.2.2实验数据处理实验数据处理是通过对原始实验数据进行一系列的物理校准和校正,最终得到与实验仪器和样品厚度等无关的,体现样品本质特征的绝对散射强度(dΣ(q)dΩ,cm-1)随着散射矢量(q,Å-1)变化的信息. 一个完整的实验通常包括5组数据的采集:空样品池透过率数据Tc(λ)、空样品池散射数据Iexpcb(q)、样品加样品池透过率数据Tsc(λ)、样品加样品池散射数据Iexpscb(q)、空背底测量Ibackground(下标s表示样品,下标c表示样品池,下标b表示背底). 小角中子散射实验中,散射信号Iexpscb(q)有以下来源:样品、样品池和各种背底(如天然背底、空气散射和电子学噪声等).各种散射信号之间的关系可以用式(1)和式(2)表示,其中I0(λ)代表零散射角度的散射强度. 扣除样品池的散射和其他各种背底,最终计算得到dΣ(q)dΩ. 式(1)和式(2)只是简化和近似,真实SANS数据处理还需要考虑探测器效率、死时间和入射中子波长分布等因素[14].2.3实验数据分析SANS数据分析方法多种多样. 一般来说,可分为不依赖于模型的分析方法和依赖于模型的分析方法. 不依赖于模型的分析方法植根于数学,是数据分析的起点. 具体来说,包括吉尼尔(Guiner)、Porod、Kratky等分析方法. Guiner分析方法是样品的散射强度的自然对数对散射矢量的平方作图,即1n(I(q))对q2作图,在qRgPorod分析方法是主要用于分析散射体尺寸的局部结构信息,要求qRg1. Porod作图即是将散射强度对散射矢量作图,即1g(I(q))对lgq作图,其斜率即为散射体的Porod因子n. 高q的散射数据通常可表示为或者对于长棒形散射体,n=1;对于二维光滑散射体,n=2;如果三维散射体拥有光滑表面,n=4; 否则,n为3~4之间. 对于大分子链,Porod因子与排斥体积参数ν有关,即n=1/v,对于稀溶液中的有排斥体积高斯链n=5/3(或者1/0.588),对于稀溶液中没有排斥体积的高斯链n=2,对于完全蹋缩的大分子链n=3.n为2~3之间可能是枝状大分子或者是形成网络结构.图3为半径为R=50 nm的硬球的散射模型,可以用贝塞尔方程拟合. 对曲线低q区域(qRg≤1)进行Guinier拟合,如图3中的小插图所示,得到均方旋转半径为38.94 nm,与理论值500 × (3/5)0.5 = 38.73 nm相符. 需要注意的是在得到 Rg之后需要进行一次验证,验证拟合区间确实满足qRg≤1.Fig. 3Guinier and Porod fit of the form factor of the hard sphere with a radius of 50 nm.对高q区域(qRg1)进行Porod拟合,得到斜率为-4.0,符合光滑球体表面分形维数. 更详细的关于Guiner、Porod和Kratky作图的图文解释和示例,读者可以参考Hammouda的SANS TOOLBox的第15章[15].常用的依赖于模型的分析方法是借助已知的样品信息,以有限多个初始参数建立正空间中散射体的几何模型,并根据公式(13)计算与之对应的倒空间的数学曲线,采用最小二乘法,不断迭代输入参数,直到模型的计算散射曲线与实验曲线的偏差在可接受范围内. 常用的分析软件有Igor[16]和SASView[17]等. Svergun和McGreevy等发展了新从头算起(ab initio)和逆蒙特卡罗模拟的分析方法[18~21],可以将正空间三维结构的傅里叶变换与散射曲线进行比较.对依赖模型的分析方法,初始模型的设计至关重要. 所以在SANS实验之前,需要进行一系列的前置散射、光谱或者成像实验,估计样品的初始结构. 根据不依赖于模型的Guinier和Porod等方法对一维散射曲线的分析结果,验证初始模型的选择是否正确. 需要注意的是,拟合参数或者基本假设越少,分析结果的准确性越高. 拟合参数多的方程可以拟合大多数SANS曲线,但必须通过结合其他研究手段固定大部分的参数.3大分子相关领域典型应用小角中子散射在物理学、化学、材料、生命科学和工业界等均有大量应用. 本文主要聚焦于大分子领域,即合成高分子、生物大分子和大分子材料领域的典型应用. 为方便讨论,依据样品的特点进行分类,分为高分子溶液、高分子共混物和复合材料、高分子结晶、凝胶、多孔材料、生物大分子. 以下就这些方面的一些经典案例和最新发现进行讨论. 由于小角中子散射应用领域众多,并且各个领域之间还会出现交叉和重叠,所以以下分类讨论并不一定严格和全面,本文只是抛砖引玉,旨在说明小角中子散射的特点和在各领域的典型应用.3.1高分子溶液体系大多数用户使用SANS研究溶液体系是为了得到溶质的多尺度形貌,所以高分子溶液体系的样品处理,实验方法,数据处理与分析具有普适性[22,23]. 大分子在溶液中的基本构象(confor-mation)的确定需要使用SANS进行证明,一般在稀溶液测定. 1974年,Cotton等使用SANS研究了线形聚苯乙烯(PS)在二硫化碳(良溶剂)和环乙烷(θ溶剂)中的构象,验证了高分子在良溶剂中是有排斥体系的高斯链,分形维数5/3,在θ溶剂中是无扰高斯链,分形维数是2[24]. 随着高分子化学的进步,科学家们合成了不同几何形状的单分散大分子. 2014年,Goossen等使用SANS研究了环形PS在氘代甲苯(良溶剂),氘代环乙烷(θ溶剂)和氘代线形PS(类θ本体)中的构象,如图4所示[25]. 环形PS在良溶剂中,Porod区间的表观分形维数1.56,小于线形PS在良溶剂中的5/3,作者解释是由于第2维利系数(A2)的影响,通过扣除A2,得到没有端基的环形PS在良溶剂中的分形维数;环形PS在θ溶剂和相同分子量的PS本体中,分形维数为2. 我们需要着重指出两点:一是对θ溶剂体系,或者高分子本体体系,图4的拟合区间在0.006~0.2 Å-1,对于低q区间,0.002 Å-1qP(q)的基本定义(公式(13))进行计算[15].Fig. 4Scattering functions and representative slopes for the overall and internal structure of ring polystyrene in good andθ solvents at different length scales. The linear polymeric matrix in the ring/linear blend is congruent with the θ-solvent. (Reprinted with permission from Ref.[25] Copyright (2015) American Chemical Society).相分离过程的研究是高分子溶液研究领域的重点之一. 大多数情形下,基于平均场理论的Ornstein-Zernike方程可以描述溶液中相分离过程的浓度涨落的变化[26,27]. Jia等使用SANS,研究了聚(N,N′-二乙基丙烯酰胺)(PDEA)在氧化三甲胺(TMAO)水溶液中的相分离发生前浓度涨落(concentration fluctuation)的变化,如图5所示[28]. 浓度涨落的强度和幅度都随温度升高而增大,随TMAO含量的增高而增大;通过外推零散射角度散射强度的倒数随着温度的倒数曲线,得到浓度涨落趋近无穷时的温度,就可以得到该共混体系的旋节线相图. 同样,这里需要注意两点:一是SANS是唯一的直接测量旋节线相图的研究手段,其他研究手段,例如浊度法,测量的都是双节线相图;二是越靠近相边界,浓度涨落的尺度越大(图5),这与温敏性高分子靠近最低共溶温度(LCST)时体积收缩[29]并不矛盾:由于图5的SANS实验的衬度来源于浓度涨落的微区,而不是单链高分子. 如果需要看到PDEA单分子链的LCST塌缩(就像使用动静态激光光散射观察PDEA极稀水溶液一样),需要使用衬度匹配技术. 典型的例子可以参考Hammouda等的实验,使用氘代和氢化聚(N-异丙基丙烯酰胺)(PNIPAM)在衬度匹配的重水/水混合溶剂中,用SANS观察PNIPAM单链的塌缩过程[30].Fig. 5SANS profiles of 4% mass fraction PDEA in TMAO-d9/D2O mixtures. (a) Temperature dependence of PDEA atcTMAO = 0.28 mol/L the arrow is used to guide the eye, indicating the increase of concentration fluctuations with temperature. (b) TMAO concentration dependence at 15 °C when TMAO concentrations are 0, 0.1, 0.28, 0.44, 0.58, 0.76, 0.90, 1.13 and 1.25 mol/L, respectively. (Reprinted with permission from Ref.[ 28] Copyright (2017) American Chemical Society).随着大分子在溶液中的浓度增加,分子之间相互作用(SI(q))逐渐变强,这时相互作用在散射曲线上将会表现为最小散射矢量附近的散射强度相对无相互作用时变小,中间q区间的散射强度相对无相互作用时变强. 如果体系中存在复杂的相互作用,如氢键相互作用、静电相互作用、憎水相互作用、π-π堆叠作用[31]等,在溶液中将形成亚稳的并且能够响应外界刺激的微相自组装结构,在污水净化、废油回收、药物输送等方面有着广泛的应用[32]. 小角中子散射是研究这类体系的非常有效的方法,既可以研究大分子或组装体在溶液中的结构(P(q))的变化[33],又可以研究组装体的结构在溶液中的相互作用(SI(q)).大分子组装结构是小角中子散射研究的一个热点. Sternhagen等合成了一系列的两亲性离子类肽嵌段共聚物,这些共聚物唯一不同的是肽链序列的离子单体的位置不同. SANS研究表明,这些肽嵌段共聚物组装成星形胶束结构,并且离子单体的位置越靠近星形胶束中心,胶束的均方旋转半径越小,并且二者呈现一定的指数关系[34]. 此项研究为利用肽键氨基酸序列调控组装胶束结构开辟了新的道路.3.2高分子共混物和复合材料通过将高分子共混、复合,石油化工工业只需要生产常见的几十种高分子材料,如聚乙烯、聚丙烯、聚酰胺等,就可以大致满足人们日常生活对高分子材料的硬度、弹性、机械强度、疲劳强度、导电性、透光性、耐热性、阻燃性、吸水性、耐酶性等多方面的需求. 这表明高分子共混物和复合材料的多相多尺度微观结构及其演化过程与宏观性能密切相关. 小角中子散射适用于实时追踪这类体系的微观结构的变化.通常非晶高分子本体或者共混物中,由于要观察的目标大分子与其周围环境的化学结构大致相同,对大部分研究手段而言衬度几乎都为0,无法看到单一高分子链或者选择性观察某一相高分子. 少部分的观察手段,包括单分子荧光或者核磁虽然有选择性地观察能力,但是前者引入了大尺寸的荧光基团,有可能影响体系的动力学和动态学行为;后者直接观察的是能量空间. 只有SANS可以通过衬度匹配具有选择性地观察单链结构的能力[35].高分子共混物在双节线相区,初级成核过程究竟是如何发生的?到现在仍然是一个非常具有挑战性的课题. Balsara课题组曾进行了深入的研究[36]. 他们使用时间分辨SANS,研究了氘代聚乙基丁烯(dPE)、聚甲基丁烯(PM)和聚(甲基乙烯-b-乙基丁烯)的三元共混物相分离初期的成核过程,如图6所示. SANS的中子束流强度低,需要较长时间(通常大于3 min,依赖于不同中子源或者SANS谱仪)才能得到满足统计误差的散射谱图. 嵌段共聚物hPM-hPE的加入是为了增强dPE/hPM的相容性,降低相分离温度并延长相分离时间,从而满足SANS采样所需时间.图6(a)表明,相分离未发生时,体系为均相,相对散射强度不随散射矢量q变化;随着相分离发生,低q散射曲线随相分离时间增长,不断向上倾斜,这说明有相分离成核的尺寸逐渐增大,零散射矢量处散射强度随之增长. 使用不依赖具体模型的Guinier方程对SANS数据进行拟合(图6(b)),可以得到零散射矢量处散射强度(In)随其均方旋转半径(Rg)变化的标度关系,分形维数1/0.54,说明初级成核也许并不是Gibbs成核过程(分形维数3),而是浓度涨落诱导过程(分形维数2).Fig. 6(a) Dependence of SANS profiles on time during the early stage of the sample with 50 vol% block copolymer. The solid lines in represent fits to the Guinier model. (b) A lg-lg plot ofRg at a given time versus In(In = I(Q=0,t)/I(Q=0,t=0)) at that time. The solid line represents the best power law fit. (Reprinted with permission from Ref.[36] Copyright (1996) The American Physical Society).复合大分子材料在工业界有着十分广泛的应用. Liu等利用小角中子散射和电子显微镜研究纳米二氧化硅球(20 nm左右)和橡胶复合体系,发现SiO2会形成24~97个硅球的聚集体,聚集体尺寸随着SiO2球体积分数增加线性变小,最佳的二氧化硅的体积分数在40%~50%之间[37,38].具有刺激响应的智能大分子材料,如自愈(self-healing)复合材料是目前研究的热点. Staropoli 等利用小角中子散射和流变实验研究靠氢键结合而成的瞬态枝化梳状大分子在熔融状态下的氢键形成机理[39]. 结果表明,瞬态链合结构对此类材料至关重要.3.3高分子结晶高分子结晶过程极为复杂,尽管科学家们进行了多年不间断地研究,一些基础性的问题仍有疑问. 1977年,Sadler等使用SANS研究了氘代聚乙烯经过溶液和熔融结晶生成的晶体内部的单链构象[40],在一系列假设下(氘代和氢化聚乙烯无相分离、同时结晶),证明了高分子单链在溶液中优先按照近邻折叠模型结晶;在熔融过程中,优先按照插线板模型结晶. 这个结果争议不大,已经写入了高分子物理的教科书. 而串晶(shish-kebab)中shish的生成机理则至今仍争议不休:究竟是高分子链的拉伸、缠结网络变形或者是壁滑导致了shish的产生?Kimata等的SANS研究使shish成核理论的研究向前迈出了关键的一步[41]. 实验观察结晶过程中分子链结构变化的关键难点还在于衬度:如何能够在shish的狭小范围内看到高分子链的结构. 如之前表2所示,X射线的衬度来源于电子云密度的差别,因此SAXS可以看到二维的大分子片晶结晶区与非晶区片层之间的电子云密度差别,从而得到片晶厚度,但是SAXS看不到一根结晶大分子链与其周围链段之间的任何差别;而常规的SANS均聚物氘代和氢化二元共混同样存在问题,它虽然提供了氘代分子与周围分子之间的衬度差别,但是也引入了结晶的氘代大分子与非晶的氘代大分子之间的衬度差别. 所以Kimata之前,科学家们没有设计出合适的可以在shish中提取分子链结构的实验方法. Kimata等使用了氘代短链(S),中等链(M)和长链(L)等规丙烯(iPP)与多分散非氘带iPP进行共混,在不同温度下进行剪切实验,用SANS观察散射图样的变化,如图7所示.图7(a)中S链的各向异性散射更加显著,温度升高到168 ℃时shish开始熔化,各向异性开始逐渐消失. Kimata等用166 ℃ 时shish刚刚开始取向的散射图样减去168 ℃或者180 ℃完全熔融的背景散射,如图7(b)所示,成功得到了d-iPP链在shish中的取向信息.图7证明了长链在shish中只起引发作用,但扩散较慢,不是shish的主体.Fig. 7(a) Temperature dependence of SANS profiles of deuterium labeled iPP during heating from 25 °C to 180 °C. The labeled fraction is denoted by S, M, and L for short D, medium D, and long D, respectively. (b) The change in SANS scattering intensity between 166 and 180 °C (left) and between 168 and 180 °C (right) for each of the three deuterium-labeled blends. (Reprinted with permission from Ref.[41] Copyright (2007) American Association for the Advancement of Science).3.4凝胶溶胶或者溶液中的胶体粒子或者大分子在合适条件下相互连接,形成空间网络结构,最后失去流动性,整个体系变成一种外观均匀,并保持一定形态的弹性半固体,这种弹性半固体称为凝胶. 凝胶在有机体的组成中占重要地位,人体内的肌肉、皮肤、细胞膜、血管壁,以及毛发、指甲、软骨等都可看作是凝胶. 相对于稀溶液,凝胶体系中的结构和相互作用更加复杂,小角中子散射方法,可用于研究此类体系的微观结构[42,43]、凝胶相的形成过程[44]和形成机理等[45].Endo等利用SANS研究不同浓度的间规聚丙烯(sPP)在氘代十氢萘溶剂中形成的物理凝胶的结构[46],散射曲线如图8(a)所示. 散射曲线在某一q范围的斜率表示在相应正空间尺度上散射体的分形维数. 浓度最低的sPP十氢萘溶液(2 wt%)的散射曲线低q区间分形维数1,说明在交联点之间有棒状结构,中等q值范围内分形维数4,类似光滑球形外表面. 所以假设sPP纳米晶为球形结构(用贝塞尔方程拟合),纳米晶之间存在的非晶sPP链形成的网络结构(用Ornstein-Zernike方程拟合),纳米晶球之间进行Percus-Yevick近似,就可以得到交联点形状、尺寸随sPP浓度和温度变化的定量关系(图8(b)).Fig. 8(a) SANS profiles of the nitrogen quenched gel with differentsPP concentrations (symbols) and corresponding fitting results (solid lines). The profiles are vertically shifted to avoid the overlap. (b) Schematic illustration of hierarchical structures in gel LN suggested by the SANS profiles. (Reprinted with permission from Ref.[46] Copyright (2019) The Royal Society of Chemistry)3.5多孔材料中子直接作用于原子核,具有很强的穿透性,可以轻松穿透较厚的多孔材料,从而在1~100 nm范围内研究其内部孔隙的孔隙率、尺寸分布、各向异性、孔的连接性和比表面积,并且可以追踪这些参数对其容纳和吸附性能的影响.Yang等利用小角中子散射研究我国四川盆地龙马溪页岩的多孔结构[47,48]. 用多分散球形孔模型和Porod方法分析中子散射数据得到的比表面积和孔隙率,都大于压汞法得到的结果,说明样品中存在盲孔. 随着样品埋藏深度的增加,盲孔数量也随之增加,并且与有机碳含量存在相关性. 这个例子需要注意样品多重散射对散射曲线的影响,通常页岩样品厚度在200 μm的情况下可以保证单次散射;具体实验中需要测量不同厚度样品散射曲线来避免多重散射.碳纤维是重要的工业材料,小角中子散射可以对碳纤维内的孔隙缺陷进行精确的表征. Jafta等利用小角中子和小角X射线对多孔碳纤维内的孔隙率和比表面积进行了精确的分析[49]. 同时还用弦长分布函数分析了体系中孔隙的空间分布,发现孔的分布相对无序. 如果多孔材料的孔隙分布比较窄,就可以用于研究液体在空间受限行为、各种气体在孔隙内的吸附和脱吸附. Melgar等利用多金属氧酸盐为水分子提供含有不同配体的孔隙,研究水分子在孔隙内的分布情况[50],研究表明,当孔隙小于1.1 nm,水分子将不能进入孔隙从而去润湿. Bahadur等利用小角中子散射研究二氧化碳在多孔碳材料内的高压吸附行为[51]. 观察到二氧化碳在微孔内随着压强的非线性吸附,微孔尺寸从约5 Å增加到7 Å. 但氩气在同样压强作用下的吸附并没有引起孔隙尺寸的变化. 说明吸附二氧化碳后,孔隙内的压强大于外界压强,推测孔内存在很强的吸附引起的溶解压.3.6生物大分子生物大分子种类丰富,多尺度结构复杂,其内部结构和作用原理的解析对解开生命的奥秘、开发新型药物等意义重大. Shi和Li对小角X射线在该领域的研究进展和一般分析方法进行了详细的阐释[52],介绍的分析方法和研究方向与本小节介绍的内容有一些类似和重叠,有兴趣的读者可以自行查阅. 中子凭借其特性和与X射线的互补在生物大分子方面的应用前景也十分广阔[53].生物大分子的小角中子散射表征难度相对较高,第一,氘代样品的制备难度大,需要利用氘水和氘带碳源培养特定的细菌,粉碎后再纯化需要的氘带样品;第二,小角中子散射是一种低空间分辨率的表征手段,对于复杂体系的散射,人们通常将小角中子散射与其他实验手段和分析方法如透射电镜、X射线晶体衍射、核磁共振以及模拟方法等结合起来对散射数据进行分析,如图9所示. David等综述了利用小角散射研究生物大分子[54]. 在生物大分子方面小角中子散射的研究内容包括但不限于:(1)肽链、核酸、蛋白质[55]、双层磷脂膜、淀粉、纤维素等生物大分子在不同环境下的结构;(2)肽链、核酸、蛋白质和双层磷脂膜等的相互作用和组合结构;(3)病毒、细胞器等.Fig. 9A scheme of an SAS experiment, structural tasks addressed and the joint use with other methods. The nominal resolution of the scattering data is indicated asd = 2 p/s. (Reprinted with permission from Ref.[56] Copyright (2007) Elsevier Ltd.).对于生物大分子这类复杂体系,在能够达成科学目标的前提下,模型设计需要尽可能地简单,将变量维持在可接受范围内. 如果散射体非常复杂,由多个具有不同结构、功能的部分组成,需要使用氘代对各个部分进行衬度匹配. 数据分析方面,第一步,对散射数据做定性或半定量的分析,例如稀溶液,可以通过Guinier作图分析散射体均方旋转半径,Porod作图分析体系拓扑结构或者分形维度;第二步,依据已知数据建立模型,分析数据. 数据分析模型通常有以下2种:第一种是依赖于散射数据的可迭代优化模型,依据模型的计算曲线和实验曲线的均方差对模型的一些变量进行迭代优化,如规则几何模型拟合、逆蒙特-卡洛(RMC)方法[21,57]、从头计算(ab initio)方法[20]等;第二种是不依赖于散射数据的独立模型(强烈依赖于所用力场),例如独立的分子动力学或者蒙特-卡洛模型,独立模型的计算SANS曲线可以与实验曲线对比,或者依据实验曲线与模型得到的可能结构进行筛选[58].限于篇幅,以下举几个有代表性的实例. 如图10为天冬氨酰-tRNA合成酶(Aspartyl-tRNA synthetase complexed)与tRNA复合物结构的小角X射线和小角中子散射联合研究图示[59]. Petoukhov和Svergun分别利用ab initial的串球模型分析复合体系的低分辨结构,如图10(A)和10(B)所示,然后利用复合物各个部分的X射线晶体学结构和刚体建模方法拟合X射线和中子散射数据,得到体系在溶液中的高分辨结构模型.Fig. 10(A) Aspartyl-tRNA synthetase complexed with tRNA. (a, b) Comparisons of the crystal structure with the ab initio bead models generated by the program MONSA. In the high resolution model, the protein and tRNA are shown as blue and magenta backbones, in the bead model corresponding phases are presented in gray and yellow, respectively. (c) Best rigid body model generated by SASREF. (d) A SASREF model with different orientations of tRNA. Right view is rotated by 90° about horizontal axis. (B) Scattering profiles from the Aspartyl-tRNA synthetase complex with tRNA. The simulated data are shown by dots, the fits obtained by the program MONSA and the program SASREF are displayed as red solid and blue dashed lines, respectively. 1 and 2 are X-ray scattering curves of the dimeric protein and the entire complex, respectively. 3-7 are neutron scattering patterns at 0, 40%, 55%, 70% and 100% D2O, respectively. The patterns are displaced in logarithmic scale for better visualization. (Reprinted with permission from Ref.[59] Copyright (2006) Springer European Biophysics Journal).同步辐射和X射线晶体学是研究生物大分子结构的利器,在得到蛋白质的晶体结构后,利用刚体建模方法,或者分子动力学模拟,结合小角X射线和小角中子散射,可以研究各类蛋白在溶液中的结构和相互作用. Shrestha等利用小角中子散射、小角X射线散和分子动力学模拟研究天然无规蛋白(intrinsically disordered protein)结构[60],发现Flory指数为0.54,介于理想链的0.5和自避行走链的0.588之间.4总结小角中子散射技术在基础、应用、产业化的各个领域中都有广泛的应用. 由于篇幅所限,本文只是首先从原理和实践两个方面对这一技术进行了简要的介绍,然后列举了小角中子散射在高分子溶液、高分子共混物和复合材料、高分子结晶、凝胶、多孔材料和生物大分子等体系结构表征方面的一些典型应用,希望能够进一步扩展我国的SANS用户群体. 如果需要更深一步了解SANS或者中子散射技术在高分子科学中的应用,可以参考一些专业书籍[12,61,62].参考文献1Borsali R,Pecora R.Soft-Mattter Characterization.Springer,2008.377-9522Cebe P,Hsiao B S,Lohse D J.Scattering from Polymers Characterization by X-rays, Neutrons, and Light.Washington DC:American Chemistry Society,2000.1-1163Roe R J.Methods of X-ray and Neutron Scattering in Polymer Science.Oxford:Oxford University Press,2000.1-804Feigin L A,Svergun D I.Structure Analysis by Small-Angle X-Ray and Neutron Scattering.New York and London:Plenum Press,1987.275-320.doi:10.1007/978-1-4757-6624-0_95Dianoux A J,Lander G.Neutron Data Booklet Second Edition (July 2003).2020-10-25.https://www.ill.eu/fileadmin/user_upload/ILL/1_About_ILL/Documentation/NeutronDataBooklet.pdf6National Nuclear Data Center.Evaluated Nuclear Data File (ENDF).2020-10-25.https://www.nndc.bnl.gov/exfor/endf00.jsp.doi:10.2172/9818137Zuo T S,Cheng H,Chen Y B,Wang F W.Chinese Phys C,2016,40(7):76204.doi:10.1088/1674-1137/40/7/0762048Carpenter J M, Agamalian M.J Phys:Conference Series,2010,251:012056.doi:10.1088/1742-6596/251/1/0120569Han Z,Zuo T,Ma C,Cheng H.Instrum Sci Technol,2019,47:448-465.doi:10.1080/10739149.2019.159773310Zhang H,Cheng H,Yuan G,Han C C,Zhang L,Li T,Wang H,Liu Y T,Chen D.Nucl Instrum Meth A2014,735:490-495.doi:10.1016/j.nima.2013.09.06511Anderson K.Reactor & Spallation Neutron Sources.Oxford:Oxford School of Neutron Scattering,2013.55-7612Higgins J S,Benoît H C.Polymers and Neutron Scattering.Oxford:Clarendon Press,1994.86-9513Rehm C,Barker J,Bouwman W G,Pynn R.J Appl Crystallogr,2013,46(2):354-364.doi:10.1107/s002188981205002914Du R,Tian H L,Zuo T S,Tang M,Yan L,Zhang J R.Instrum Sci Technol,2017,45(5):541-557.doi:10.1080/10739149.2016.127822915Hammouda B.Probing Nanoscale Structures-The SANS Toolbox.Gaithersburg:National Institute of Standards and Technology Center for Neutron Research,2010.31-19116Kline S.J Appl Crystallogr,2006,39(6):895-900.doi:10.1107/s002188980603505917Butler P,Doucet M,Jackson A,King S.SasView for Small Angle Scattering Analysis (July 2020).2020-10-25.https://www.sasview.org/18Konarev P V,Svergun D I.IUCrJ,2018,5(Pt 4):402-409.doi:10.1107/s205225251800590019Petoukhov M V,Svergun D I.Acta Crystallogr D Biol Crystallogr,2015,71(Pt 5):1051-1058.doi:10.1107/s139900471500257620Volkov V,Svergun D.J Appl Crystallogr,2003,36:860-864.doi:10.1107/s002188980300026821Gereben O,Pusztai L,McGreevy R L.J Phys Condens Matter,2010,22(40):404216.doi:10.1088/0953-8984/22/40/40421622Li Z,Cheng H,Li J,Hao J,Zhang L,Hammouda B,Han C C.J Phys Chem B,2011,115(24):7887-7895.doi:10.1021/jp203777g23Hu W T,Yang H,He C,Hu H Q.Chinese J Polym Sci,2017,35(9):1156-1164.doi:10.1007/s10118-017-1969-724Cotton J P,Decker D,Benoit H,Farnoux B,Higgins J,Jannink G,Ober R,Picot C,des Cloizeaux J.Macromolecules,1974,7(6):863-872.doi:10.1021/ma60042a03325Goossen S,Bras A R,Pyckhout-Hintzen W,Wischnewski A,Richter D,Rubinstein M,Roovers J,Lutz P J,Jeong Y,Chang T,Vlassopoulos D.Macromolecules,2015,48(5):1598-1605.doi:10.1021/ma502518p26Hao J,Cheng H,Butler P,Zhang L,Han C C.J Chem Phys,2010,132(15):154902.doi:10.1063/1.338117727Hore M J A,Hammouda B,Li Y,Cheng H.Macromolecules,2013,46(19):7894-7901.doi:10.1021/ma401665h28Jia D,Muthukumar M,Cheng H,Han C C,Hammouda B.Macromolecules,2017,50(18):7291-7298.doi:10.1021/acs.macromol.7b0150229Cheng H,Wu C,Winnik M A.Macromolecules,2004,37(13):5127-5129.doi:10.1021/ma049620130Hammouda B,Jia D,Cheng H. OAJoST,2015,3:101152.doi:10.11131/2015/10115231Datta S,Kato Y,Higashiharaguchi S,Aratsu K,Isobe A,Saito T,Prabhu D D,Kitamoto Y,Hollamby M J,Smith A J,Dagleish R,Mahmoudi N,Pesce L,Perego C,Pavan G M,Yagai S.Nature,2020,583(7816):400-405.doi:10.1038/s41586-020-2445-z32Zhang H V,Polzer F,Haider M J,Tian Y,Villegas J A,Kiick K L,Pochan D J,Saven J G.Sci Adv,2016,2(9):e1600307.doi:10.1126/sciadv.160030733Wang Z,Faraone A,Yin P,Porcar L,Liu Y,Do C,Hong K,Chen W R.ACS Macro Lett,2019,8(11):1467-1473.doi:10.1021/acsmacrolett.9b0061734Sternhagen G L,Gupta S,Zhang Y,John V,Schneider G J,Zhang D.J Am Chem Soc,2018,140(11):4100-4109.doi:10.1021/jacs.8b0046135Zuo T,Ma C,Jiao G,Han Z,Xiao S,Liang H,Hong L,Bowron D,Soper A,Han C C,Cheng H.Macromolecules,2019,52(2):457-464.doi:10.1021/acs.macromol.8b0219636Balsara N P,Lin C,Hammouda B.Phys Rev Lett,1996,77(18):3847-3850.doi:10.1103/physrevlett.77.384737Liu D,Song L,Song H,Chen J,Tian Q,Chen L,Sun L,Lu A,Huang C,Sun G.Compos Sci Technol,2018,165:373-379.doi:10.1016/j.compscitech.2018.07.02438Liu D,Chen J,Song L,Lu A,Wang Y,Sun G.Polymer,2017,120:155-163.doi:10.1016/j.polymer.2017.05.06439Staropoli M,Raba A,Hövelmann C H,Krutyeva M,Allgaier J,Appavou M S,Keiderling U,Stadler F J,Pyckhout-Hintzen W,Wischnewski A,Richter D.Macromolecules,2016,49(15):5692-5703.doi:10.1021/acs.macromol.6b0097840Sadler D M,Keller A.Macromolecules,1977,10(5):1128-1140.doi:10.1021/ma60059a04541Kimata S,Sakurai T,Nozue Y,Kasahara T,Yamaguchi N,Karino T,Shibayama M,Kornfield J A.Science,2007,316(5827):1014.doi:10.1126/science.114013242Shibayama M,Li X,Sakai T.Colloid Polym Sci,2018,297:1-12.doi:10.1007/s00396-018-4423-743Gao J,Tang C,Elsawy M A,Smith A M,Miller A F,Saiani A.Biomacromolecules,2017,18(3):826-834.doi:10.1021/acs.biomac.6b0169344Srivastava S,Andreev M,Levi A E,Goldfeld D J,Mao J,Heller W T,Prabhu V M,de Pablo J J,Tirrell M V.Nat Commun,2017,8:14131.doi:10.1038/ncomms1413145Nishi K,Fujii K,Katsumoto Y,Sakai T,Shibayama M.Macromolecules,2014,47(10):3274-3281.doi:10.1021/ma500662j46Endo F,Kurokawa N,Tanimoto K,Iwase H,Maeda T,Hotta A.Soft Matter,2019,15(27):5521-5528.doi:10.1039/c9sm00582j47Yang R,He S,Hu Q,Sun M,Hu D,Yi J.Fuel,2017,197:91-99.doi:10.1016/j.fuel.2017.02.00548Sun M,Yu B,Hu Q,Zhang Y,Li B,Yang R,Melnichenko Y B,Cheng G.Int J Coal Geology,2017,171:61-68.doi:10.1016/j.coal.2016.12.00449Jafta C J,Petzold A,Risse S,Clemens D,Wallacher D,Goerigk G,Ballauff M.Carbon,2017,123:440-447.doi:10.1016/j.carbon.2017.07.04650Melgar D,Zhou Q,Chakraborty S,Porcar L,Weinstock I A,Ávalos J B,Wu B,Bo C,Yin P.J Phys Chem C,2020,124(18):10201-10208.doi:10.1021/acs.jpcc.0c0101951Bahadur J,Melnichenko Y B,He L,Contescu C I,Gallego N C,Carmichael J R.Carbon,2015,95:535-544.doi:10.1016/j.carbon.2015.08.01052Shi Ce(史册),Li Yunqi(李云琦).Acta Polymerica Sinica(高分子学报),2015, (8):871-883.doi:10.11777/j.issn1000-3304.2015.1504853Fitter J,Gutberlet T,Katsaras J.Neutron Scattering in Biology: Techniques and Applications.Berlin Heidelberg and New York:Springer,2006.doi:10.1007/3-540-29111-354Jacques D A,Trewhella J.Protein Sci,2010,19(4):642-657.doi:10.1002/pro.35155Koruza K,Lafumat B,ÁVégvári,Knecht W,Fisher S Z.Arch Biochem Biophys,2018,645:26-33.doi:10.1016/j.abb.2018.03.00856Petoukhov M V,Svergun D I.Curr Opin Struct Biol,2007,17(5):562-571.doi:10.1016/j.sbi.2007.06.00957Ma Chang-li(马长利),Cheng He(程贺),Zuo Taisen(左太森),Jiao Guisheng(焦贵省),Han Zehua(韩泽华),Qin Hong(秦虹).Chinese Journal of Chemical Physics(化学物理学报),2020,33(6s):727-732.doi:10.1063/1674-0068/cjcp200507758Jiao G,Zuo T,Ma C,Han Z,Zhang J,Chen Y,Zhao J,Cheng H,Han C C.Macromolecules,2020,53(13):5140-5146.doi:10.1021/acs.macromol.0c0078859Petoukhov M V,Svergun D I.Eur Biophys J,2006,35(7):567-576.doi:10.1007/s00249-006-0063-960Shrestha U R,Juneja P,Zhang Q,Gurumoorthy V,Borreguero J M,Urban V,Cheng X,Pingali S V,Smith J C,O’Neill H M,Petridis L.Proc Natl Acad Sci,2019,116(41):20446-20452.doi:10.1073/pnas.190725111661Han C C,Akcasu A Z.Scattering and Dynamics of Polymers: Seeking Order in Disordered Systems.Singapore:John Wiley & Sons (Asia) Pte Ltd,2011.1-98.doi:10.1002/978047082484962Zemb T,NeutronLindner P.X-rays and Light.Scattering Methods Applied to Soft Condensed Matter.Amsterdam:Elsevier,2002.1-552.doi:10.1107/s0021889803001808原文链接:http://www.gfzxb.org/thesisDetails#10.11777/j.issn1000-3304.2020.20242&lang=zhDOI:10.11777/j.issn1000-3304.2020.20242《高分子学报》高分子表征技术专题链接:http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304






















 我要推广仪器
我要推广仪器
 下载APP
下载APP