请问有用氨基柱检测肾上腺素、去甲肾上腺素、多巴胺的吗氨基柱可以检测这些极性大的生物碱吗
请问有人用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测过血浆里面肾上腺素、去甲肾上腺素或者多巴胺其中的一种吗本人尝试好多方法都没检测出来,想请教一下谢谢了
复方盐酸阿替卡因注射液为复方制剂,是盐酸阿替卡因与肾上腺素的灭菌水溶液,作为口腔用局部麻醉剂,适用于涉及切骨术及粘膜切开的外科手术过程。[img=,144,61]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191545418661_4518_2222981_3.jpg!w144x61.jpg[/img][color=black] [/color][color=#3e3e3e]肾上腺素 L(-)-Epinephrine M.W. : 183.2 [/color][b]在国家药品标准(YBH17082004-2015Z)[/b]中,在对复方盐酸阿替卡因注射液中肾上腺素进行分析时,使用[b]甲醇-水[/b]进行梯度洗脱,但由于[b]肾上腺素极性较强[/b],即使初始梯度为纯水相条件,肾上腺素仍紧邻死时间出峰,[b]保留不佳,易受到溶剂峰干扰,无法进行准确定量。[/b]我们分别尝试使用反相柱CAPCELL PAK C18 MGII加离子对试剂,以及直接使用离子交换色谱柱CAPCELL PAK SCX UG80两种方式,对复方盐酸阿替卡因注射液中肾上腺素和硫酸肾上腺素进行保留分析(复方盐酸阿替卡因注射液由客户提供)。CAPCELL PAK C18 MGII液相色谱柱,其采用高纯度硅胶作为基质,通过减少硅胶微细孔的数量来增大有效比表面积;并且采用新包被技术Ultimate Polymer Coating,实现了对硅醇基极大程度的封锁,兼具分离性能和普适性能,通用性非常好。CAPCELL PAK SCX UG80是强阳离子交换柱,使用高纯度硅胶,填料中金属杂质很少,使配位化合物的吸附得到了极大程度抑制,兼具聚合物和硅胶填料的优点。[b][color=#0070c0]实验方法[/color][color=#0070c0]方法一[/color][color=#0070c0]使用[/color][color=#0070c0]CAPCELL PAK C18MGII[/color][color=#0070c0]色谱柱[/color][color=#0070c0]+[/color][color=#0070c0]离子对试剂[/color][/b]如图1,对肾上腺素对照品溶液进行分析,肾上腺素主峰保留时间为5.69 min,拖尾因子为1.19,理论塔板数为12538。在相同色谱条件下,尝试对亚硫酸肾上腺素标准品及注射液中的亚硫酸肾上腺素进行分析。如图3,亚硫酸肾上腺素标准品溶液能够得到良好分析结果,注射液(客户提供的样品)中未明显见亚硫酸肾上腺素出峰,保留时间为3.55min,拖尾因子为1.14,理论塔板数为14955。[b][color=#0070c0]方法二[/color][color=#0070c0] CAPCELL PAK SCX UG80[/color][color=#0070c0]色谱柱[/color][/b][color=#000000]考虑到使用离子对试剂的流动相条件具有流动相配制麻烦、有损色谱柱寿命、平衡时间长等缺点,我们也尝试使用键合磺酸基团的强阳离子交换柱 ——CAPCELL PAK SCX UG80进行分析。[/color][color=#000000]如图4,在流动相中添加磷酸二氢铵,通过对盐浓度进行调整,在5 mmol/L磷酸二氢铵(磷酸调pH=2.5)条件下,亚硫酸肾上腺素保留时间为3.32 min,然而出现峰形拖尾现象,拖尾因子为2.0,不如CAPCELL PAK C18 MGII色谱柱添加离子对试剂所得分析结果好。[/color][align=center][/align][align=left][img=,400,284]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547381421_926_2222981_3.jpg!w584x416.jpg[/img] [img=,400,276]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548310561_8067_2222981_3.jpg!w572x395.jpg[/img][/align][align=left][img=,400,166]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547576748_522_2222981_3.jpg!w612x254.jpg[/img] [img=,400,167]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548525761_4184_2222981_3.jpg!w624x262.jpg[/img][/align][align=left]图1 MGII分析肾上腺素对照品溶液结果(离子对条件) 图2 MGII分析注射液结果(离子对条件)[/align][align=center][/align][img=,400,258]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191550354951_2539_2222981_3.jpg!w644x416.jpg[/img] [img=,400,250]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191555486850_3396_2222981_3.jpg!w644x403.jpg[/img][img=,400,147]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191551260881_9828_2222981_3.jpg!w696x256.jpg[/img] [img=,400,164]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191556163846_9739_2222981_3.jpg!w632x260.jpg[/img][align=left]图3 MGII分析亚硫酸肾上腺素标准品和注射液结果(离子对条件) 图4 SCX UG80分析亚硫酸肾上腺素对照品溶液和供试品溶液[/align][align=left][/align][align=left]综上实验结果,使用中等极性色谱柱CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 250 mm,在流动相中添加5 mM辛烷磺酸钠、30°C柱温条件下进行梯度洗脱,能够实现复方盐酸阿替卡因注射液中肾上腺素和亚硫酸肾上腺素的良好保留与分析。[/align][b][color=#0070c0][/color][/b][align=left][b][color=#0070c0] [/color][color=#0070c0] [/color][/b][/align]
用液质做去甲肾上腺素,优化液相方法的时候,不管怎么改变水相的比例,他的出峰时间都在1min左右,做样品的时候,正好在这里有一个倒峰,干扰很严重,导致定性都有问题。我用的是thermo的C18柱。去甲肾上腺素是正离子模式。请教各位大侠,有什么好方法没,把出峰时间推迟一点。
请问一下我检测的都是标准品,为啥肾上腺素与多巴胺会出现两个锋,之前没换柱子走出来一个峰还有酪氨酸基线为什么呈波浪状这些都是标准品 自己现在刚接触[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url] 所以不知道怎么回事谢谢了[img=,149,198]https://ng1.17img.cn/bbsfiles/images/2018/11/201811092107209705_8638_3506169_3.png!w149x198.jpg[/img]
如题,我最近在用三重四级杆做维生素类化合物测定,配的是ESI源,但是在优化离子时,有些化合物子离子碎片与常规报道的对不上,本底也高,且响应很低,想问下各位大神,如果换成APCI源会不会更好一些呢?或者还会有什么其他原因导致这种情况出现呢?
山葡萄酒中多酚类化合物酚类化合物是葡萄酒中的重要生理活性物质,对人体的健康起着重要保健作用。山葡萄酒中的多酚类化合物主要有:花色苷:是一种红色素化合物,有花青素、甲基花青素、牵牛花素、锦葵花素、花翠素、芍药素、栎皮黄素等,其含量是一般葡萄酒的2倍。黄酮类:是一种黄色素化合物,有堪非醇、槲皮素、山奈酚、杨梅素等,其黄铜醇的含量为1.43g/L,是一般葡萄酒的5~10倍。儿茶素类:主要有儿茶素、表儿茶素、表没食子儿茶素等,具有一定的苔味。原花色素类:主要有原花青素、原花翠素、原天竺葵素等。是葡萄籽与皮的主要成份,也是葡萄酒中多酚类化合物含量最多的一类。单宁类:是由花白素的多聚体组成的,有一定的涩味,具有重要的生理功能。山葡萄酒中单宁的含量是一般葡萄酒的2~3倍。白藜芦醇化合物:主要有顺式白藜芦醇、反式白藜芦醇、顺式白藜芦醇糖苷、反式白藜芦醇糖苷、顺式反式白藜芦醇异构体等。这些化合物主要来源于葡萄皮、籽中,是植物体具抗病毒的生理活性物质,也是对人体防治心脑血管疾病的重要药理成份。山葡萄酒中白藜芦醇的含量为5.86~8.20mg/L,高于国际标准,是一般葡萄酒的4~6倍。多酚类化合物是重要的保健功能成份,主要来源葡萄皮、籽中,因此吃葡萄带皮、籽一起吃掉是最有益身体健康的。酶类化合物:主要有超氧化物岐化酶(SOD),是一种自由基清除剂,具有破坏活性氧作用的自卫酶类化合物。山葡萄酒中含量为1.52×104—1.84×104mg/L,虽然含量极微小,但对人体健康有重要作用,也是其它葡萄中不具备的。
酚类化合物水溶性强,一般分离后极性较大又都在水部位,比如10%甲醇请问除了挥掉甲醇后冻干这个方法外,还有没有除水的方法。因为温度一高就变黄了,所以不方便用悬蒸。

维权声明:本文为chuanxinlian原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。浅谈甾体类化合物的分离与结构鉴定 在植物环己烷或石油醚萃取部分,有很多小极性的化合物,并且这些化合物之间的极性差异也很小,分离时可能会遇到很多困难,即使分离得到了单体化合物,结构解析时也是一件很不容易的事,不管是氢谱还是碳谱中,都在高场区出来一堆信号,有时氢谱中的积分都不一定能积准,碳谱的高场区就像个小树林,密密麻麻的全是信号,看着就头疼。由于我曾从一植物的环己烷层中分离得到了多个甾体类化合物(主要为豆甾类),下面就我在实验过程中在分离和解析甾体类化合物的一点经验跟大家分享一下。1.甾体类化合物的分离纯化 由于我做的这一部分的成分极性很小,其中的甾体类化合物均不连糖,并且我所得到的化合物的结构很相似,有的只相差一个双键或只差一个羟基或羰基,总之,结构类似,极性也就相差不大,分离也就不容易了。由于极性小,我采用的分离手段只能局限在硅胶柱或板、凝胶柱(Sephadex LH-20)。首先把粗浸膏过一遍大的硅胶柱,已达到按照极性大致砍一下段的效果,由于环己烷萃取部分有很多色素,所以接下来采用过Sephadex LH-20柱将其中的色素除去,最后得到多种没有色素的流分,这里面的成分也是很杂的,有多种类型的化合物,如黄酮类、二萜内酯类、甾体类等,由于各类型化合物间的分子量有很大的差异,接下来就可以过一个又细又长的Sephadex LH-20柱,流速一定不能太快,并且流分要接的很细,我们一般用5ml的小瓶接,尽量避免不必要的交叉。这样就有可能将甾体类化合物与其他类型的化合物分开了,最后一步,在硅胶薄层板上看一下,如果成分简单,分离度良好,就采用制备薄层色谱,将单体化合物得到。 需要指出的是,制备薄层时,由于很多甾体类化合物在紫外灯下没有任何紫外吸收,这时千万不要认为没有紫外吸收的地方就没有化合物。比如这块板上的: http://ng1.17img.cn/bbsfiles/images/2010/10/201010220007_252894_1745326_3.jpg 此时,一定要在点小硅胶板时用通用显色剂显色,确定哪里有点,该流分总共有几个点,各个点之间的上下关系,有没有紫外吸收之类的,分析清楚了,最后再去做制备薄层,如果有没有紫外吸收的点,在刮板前可以先用玻璃或报纸将板的大部分挡住,在板的另一边显色,找到该点的具体位置,以免漏掉重要的点。2.豆甾类化合物的结构解析 因为我分到的甾体类化合物主要为豆甾类,所以我就只介绍此类化合物的结构解析了。下面是我在解析过程中总结的该类化合物的谱学上的特点:2.1 氢谱中最高场的信号为18-CH3,化学位移一般在0.7ppm左右。2.2 碳谱中最高场的信号同样为18-CH3,化学位移一般在12.0ppm左右。实例列举:http://ng1.17img.cn/bbsfiles/images/2010/10/201010220010_252897_1745326_3.jpg1H-NMRhttp://ng1.17img.cn/bbsfiles/images/2010/10/201010220011_252898_1745326_3.jpg13C-NMRhttp://ng1.17img.cn/bbsfiles/images/2010/10/201010220011_252899_1745326_3.jpg2.3 甾体母核上的羟基的取向不同,与其所连的碳的化学位移也会有所不同,一般情况下,α-OH连接的碳化学位移一般小于70.0ppm,而β-OH连接的碳化学位移一般大于70.0ppm。举例如下:http://ng1.17img.cn/bbsfiles/images/2010/10/201010220011_252900_1745326_3.jpg2.4 如果你拿到一套新的谱图,看到高场区有很多聚集在一起的碳氢信号,并且氢碳谱中最高场的化学位移值分别在12.0和0.7ppm左右,在你不确定它什么类型的化合物的时候,可以考虑一下是不是甾体类化合物(三萜类最高场的碳信号在18ppm左右,基本上没有12ppm左右的碳信号)。最后,希望我的原创能给某些人起到抛砖引玉的作用。
有没有大神做车内空气全谱分析或者车内硫类和胺类化合物测试的??而且是用热脱附GCMS做的??想咨询这方面的问题,私信我,有偿咨询!!!
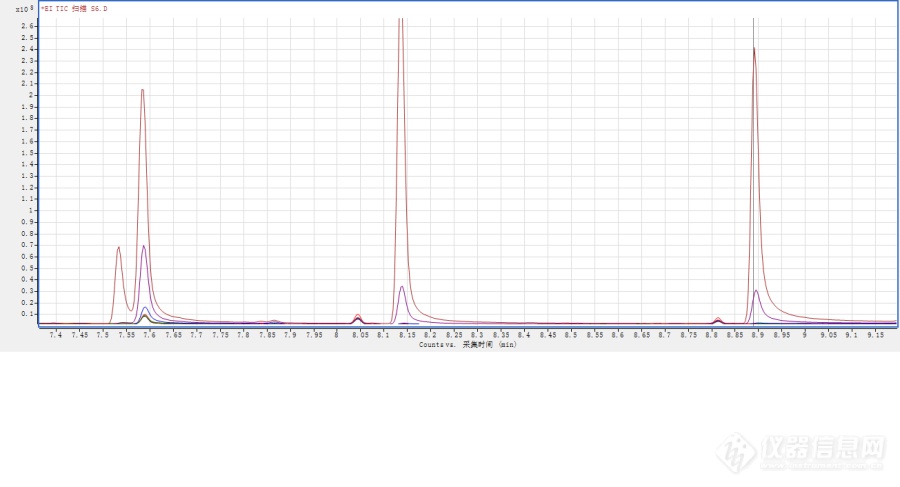
求助大神,用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]测酚类化合物 2,4-二硝基酚、4-硝基酚、2-甲基-4,6-二硝基酚、五氯酚这几种物质在[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]上一开始可以正常出峰,但是响应衰减的很快,标曲不成线性(下面图片中的四个峰是一条标曲五个点的色谱图,只有最高点明显,倒数第二个就衰减很多了);后来再打干脆不出峰了;紧接着我换了新的隔垫、衬管,切了一段色谱柱,清洗了离子源,还是不行[img]https://simg.instrument.com.cn/bbs/images/brow/em63.gif[/img]。参考HJ744用衍生化的方法,但是打出来的色谱图上根本提取不到衍生后的目标离子,一开始衍生溶液没有完全浸入水浴,后来换了顶空瓶,全部浸入水浴。难道是氮吹浓缩的时候流速太快吹飞了吗?还有个现象,酚类化合物的标准品放在室温时间长了会变黄,时间越久越黄,是标准品变质了吗(刚从冰箱取出来的时候没有颜色)?[img=,690,373]https://ng1.17img.cn/bbsfiles/images/2020/04/202004101650128460_4443_3471664_3.png!w690x373.jpg[/img]
我现在想分离黄铜类化合物,包括金丝桃苷,槲皮素,芦丁,山奈酚,看有文献缓冲液用的是硼砂和磷酸二氢钠的混合溶液,这种缓冲液如何调节PH啊,还有二者的比例对分离度有何影响啊,大家有做过的分享一下经验
近日,PLoS ONE在线发表了中科院上海生命科学研究院营养所王慧研究组的研究论文Dihydroartemisinin exerts its anticanceractivity through depleting cellular iron viatransferrin receptor-1。该研究揭示了青蒿素类化合物双氢青蒿素抗肿瘤的又一新机制。 随着我国科学家凭借对青蒿素类药物的研究获得2011年国际拉斯科临床医学大奖,青蒿素及其衍生物再度成为举世瞩目的明星分子,其中双氢青蒿素(DHA)是青蒿素的主要代谢产物和活性最强的一种衍生物。近年来,王慧研究组对青蒿素类化合物的抗肿瘤功能及机制开展了多项研究,已发现青蒿素及其衍生物对卵巢癌、肝癌的抗癌效果并探讨了其作用机制。 本研究中,该组巴乾等研究人员发现,DHA能造成肿瘤细胞铁元素的缺乏、降低铁元素的吸收、干扰细胞内铁元素既有的平衡状态,且这种改变与氧化损伤无关。进一步研究发现,DHA可以降低细胞膜上的转铁蛋白受体1(TfR1)水平,通过脂筏介导的内吞作用对其进行调控,减弱了细胞对铁的吸收,从而杀伤肿瘤细胞。 该研究结果将为青蒿素类以及铁元素靶向类抗肿瘤药物的开发提供理论基础。 该研究课题得到了国家自然科学基金委、中国科学院和上海市科委的资助。 论文链接
有没有人用HP-17的柱子做酚类化合物的
各位大神酚类化合物前处理都怎么做的?索氏提取器坐土壤中的酚类化合物条件是多少合适?HJ703-2014这个标准,净化那一步怎么我做的有机相在上层,标准是说在下层,求指点,最好能细致点的
[b][size=4] 一、性状[/size][/b][size=4] 醌类化合物随着助色团酚羟基的引入而表现出一定的颜色。引入的助色团越多,颜色则越深。[/size][size=4] [b] 二、升华性[/b][/size][size=4] 游离的醌类多具升华性,小分子的苯醌类及萘醌类具有挥发性。[/size][b][size=4] 三、溶解性[/size][/b][size=4] 游离醌类多溶于有机溶剂,微溶或不溶于水。而醌类成苷后,极性增大。[/size][b][size=4] 四、酸碱性[/size][/b][size=4] 蒽醌类衍生物酸性强弱的排列顺序为:含COOH>含二个以上β-OH>含一个β-OH>含二个以上α-OH>含一个α-OH.在分离工作中,常采取碱梯度萃取法来分离蒽醌类化合物。用碱性不同的水溶液(5%碳酸氢钠溶液、5%碳酸钠溶液、1%氢氧化钠溶液、5%氢氧化钠溶液)依次提取,其结果为酸性较强的化合物(含COOH或二个β-OH)被碳酸氢钠提出;酸性较弱的化合物(含一个β-OH)被碳酸钠提出;酸性更弱的化合物(含二个或多个α-OH)只能被1%氢氧化钠提出;酸性最弱的化合物(含一个α-OH)则只能溶于5%氢氧化钠。[/size][b][size=4] 五、显色反应[/size][/b][size=4] (1)Feigl反应 醌类衍生物在碱性条件下加热与醛类、邻二硝基苯反应,生成紫色化合物。医学教育网搜集整理[/size][size=4] (2)无色亚甲蓝显色试验 无色亚甲蓝乙醇溶液(1mg/ml)专用于检识苯醌及萘醌。样品在白色背景下呈现出蓝色斑点,可与蒽醌类区别。[/size][size=4] (3)Borntrager's反应 在碱性溶液中,羟基醌类颜色改变并加深,多呈橙、红、紫红及蓝色,如羟基蒽醌类化合物遇碱显红至紫红色,称之为Borntrager's反应。蒽酚、蒽酮、二蒽酮类化合物需氧化形成羟基蒽醌后才能呈色,其机理是形成了共轭体系。[/size][size=4] (4)Kesting-Craven反应 当苯醌及萘醌类化合物的醌环上有未被取代的位置时,在碱性条件下与含活性次甲基试剂,如乙酰乙酸酯、丙二酸酯反应,呈蓝绿色或蓝紫色。蒽醌类化合物因不含有未取代的醌环,故不发生该反应,可用于与苯醌及萘醌类化合物区别。[/size][size=4] (5)与金属离子的反应 蒽醌类化合物如具有α-酚羟基或邻二酚羟基,则可与Pb[sup]2+[/sup]、Mg[sup]2+[/sup]等金属离子形成络合物。[/size][size=4] 与Pb[sup]2+[/sup]形成的络合物在一定pH条件下能沉淀析出,与Mg[sup]2+[/sup]形成的络合物具有一定的颜色,可用于鉴别。如果母核上只有1个α-OH或1个β-OH,或2个-0H不在同环上,则显橙黄至橙色;如已有1个α-OH,并另有1个-0H在邻位则显蓝至蓝紫色,若在间位则显橙红至红色,在对位则显紫红至紫色。[/size]
各位大侠,大家做过酚类化合物吗?我的标准曲线做不出来,求救了!你们的标准曲线是什么样的?
葡萄酒酚类化合物分为两大类:非类黄酮(羟基肉桂酸、羟基苯甲酸和二苯乙烯)和类黄酮(花青素、黄酮-3-醇和黄酮醇)。这些次生代谢产物对葡萄酒的苦味、色泽、涩味、香气等主要感官参数都有重要影响,而这些都是影响消费者接受度和喜好度的最重要因素。许多研究表明,影响葡萄酒中酚类化合物存在的主要因素之一是葡萄品种。因此,用特定的葡萄品种酿造的葡萄酒通常是根据感官品质来描述的,这至少可以部分反映其品种来源。
我现在用液相色谱做水中酚类化合物的测定,标准曲线做完了,请问大家方法的检出限应该怎么计算呀?谢谢啦``
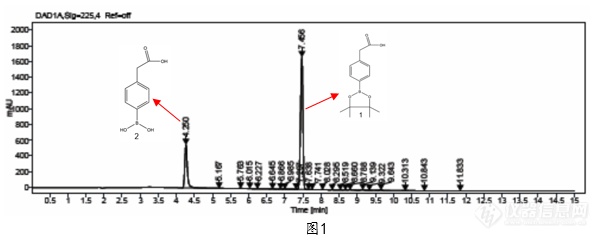
[align=left]近期我们遇到了一种硼酸酯类的化合物1,采用实验室通用方法进行检测的时候发现会出现一个很大的杂质2,根据工艺分析不可能会出现这么大的杂质,定量核磁检测发现该物质含量比较高,并不存在这个大的杂质,用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LCMS[/color][/url]去鉴定后发现该杂质为该化合物的水解杂质2(如图1)[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922409286_9676_5310417_3.png[/img][/align][align=center]图 1:流动相A: 0.05%TFA 流动相B: ACN条件下的样品色谱图[/align]为此我们判定肯定是检测方法出现了问题,首先我们排除稀释剂的影响,稀释剂为乙腈,做了相应的稳定性实验,发现临用新配情况下该杂质仍旧很大。由此我们判断可能是流动相导致该化合物1不稳定会水解生成杂质2。考虑到硼酸酯类化合物可能对酸不稳定,在酸性条件下会被催化水解成硼酸类化合物和相应的醇,因此打算更换其他流动相。首先我们尝试了碱性体系(如图2),由于该化合物1为酸性化合物,在碱性条件下保留较弱,但是从图谱可以看出水解杂质仍旧比较大,由此可以判断在碱性条件下该化合物1也并不稳定。[align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922406680_5272_5310417_3.png[/img][/align][align=center]图 2:流动相A: 0.1%NH4OH 流动相B: ACN条件下的样品色谱图[/align][align=left]随后我们又尝试了中性体系,采用中性体系的流动相进行测试(如图3)。从图3(a)可以看出,水做流动相条件下,由于流动相的离子强度不够导致峰形丑,还可以看出水解杂质2仍旧存在,但从(b)中可以看出当用乙酸铵作为流动相时候,峰形对称,水解杂质2也比较小。[/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922412671_1242_5310417_3.png[/img][/align][align=left][/align][align=left][img]https://ng1.17img.cn/bbsfiles/images/2022/11/202211090922413707_3568_5310417_3.png[/img][/align][align=center]图 3:(a)流动相A: 水 流动相B: 乙腈 (b)流动相A: 10mM 乙酸铵水溶液 流动相B: 乙腈条件下的样品色谱图[/align][align=left]根据以上结果我们猜测:该化合物对酸碱都不稳定,但中性条件下只在乙酸铵体系下稳定,为此我们从化合物1本身及水解杂质2的结构分析,该化合物1中的硼原子为sp2杂化,还存在一个空的p轨道,这个空轨道易于接受水和醇等带有未共用电子对的亲核试剂的进攻而使硼酸酯水解([font='adobeheitistd-regular'][size=13px]其机理见方程式[/size][/font][font='dlf-32769-4-2073904376+zipdfa-8'][size=13px]([/size][/font][font='dlf-3-0-25052658+zipdfa-84']1[/font][font='dlf-32769-4-2073904376+zipdfa-8'][size=13px]))。[/size][/font]继续与水作用,生成相应的醇和硼酸。[/align][align=left][/align][img]" style="max-width: 100% max-height: 100% [/img]通过对此分析,似乎已经能够解释化合物1对碱不稳定的原因,即羟基中氧上的孤对电子会进攻硼的空轨道导致其水解,至于为什么在乙酸铵体系中是稳定的我们推测原因是乙酸铵的氮原子会与硼原子形成配对键,从而使该化合物1稳定。 虽然只是硼酸酯类化合物中的一种物质的检测,但是根据检测结果和分析可以为以后的该类化合物的方法开发提供思路,即通在对硼酸酯类的化合物进行方法开发时候,尽量不要采用酸碱体系的流动相,可以考虑用乙酸铵缓冲液作为流动相进行检测。[align=left] [/align]
本人参照GBZ/T 160.72-2007标准在做工作场所硝基苯类化合物检测时,由于使用的是ECD检测器,它的响应值不稳定,作出的标准曲线的线性很差,标准上的线性范围很窄,而且职业卫生的限值并不包括在内。有哪位同仁可以传授下经验,谢谢!!
酚类化合物的结果要怎么算?测的浓度是mg/l,结果是mg/kg,还要怎么做,标准上这个试样质量是取样的量?定容体积是哪一步的?干物质质量又是啥[img=,690,920]https://ng1.17img.cn/bbsfiles/images/2019/06/201906080805311143_8582_3925306_3.png[/img]
如果要分析一类化合物 比如塑化剂 ,总共有18种化合物, 用GC-MS,刚拿到手怎么确定程序升温的过程,是不是要每一个化合物要单独进一针,确定其保留时间,离子碎片啊
想问大家个问题,就是现在我在做硝基苯类化合物,做出来的线性前面几种的线性都挺好的,后面的化合物线性都不太好。上机的方法也是按照标准来的,衬管,隔垫啥的都换过一遍了,线性还是不行,不知道是什么原因?
最近做一个三嗪二酮类化合物的检测,总有一个杂质峰不稳定,怀疑发生了烯醇互变,请教大家有没有什么好的[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]方法检测类似化合物的
在实际工作中我们会遇到这类化合物如,对甲基环己甲酸,对乙基环己甲酸等等,这类化合物都存在异构体,感兴趣的朋友,请交流下这类化合物分析的心得!
做水质氯苯类化合物hj621-2011。gc-ecd。DB-WAX柱子。色谱条件方法上的用过。标准物质证书上的色谱条件也用过。都不行。标准物质是甲醇:甲苯9:1。12种混标。用甲醇稀释做曲线。用曲线最高点氯苯的浓度是5000微克每升。目标峰很低。还没有溶剂峰高。请教大神是什么原因呢。
固体污染源排气中酚类化合物的测定四氨基安替比林分光光度法中做标准曲线时为什么吸光值会越来越小,请各位同仁前辈解惑
炔类化合物三键上氢的化学位移为什么会因氘代试剂的不同而改变?
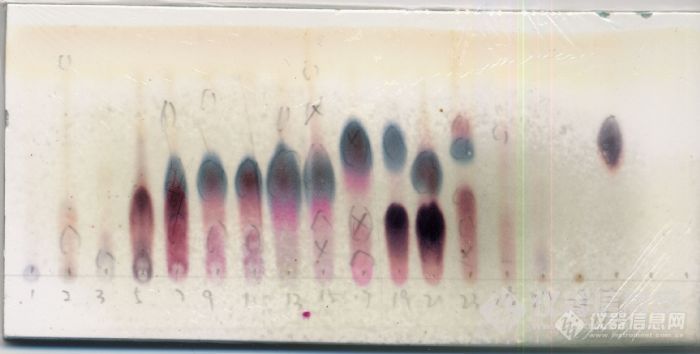
维权声明:本文为环烯醚萜原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。环烯醚萜类化合物分离纯化心得体会基本介绍 环烯醚萜(iridoids)为臭蚁二醛(iridodial)的缩醛衍生物。臭蚁二醛是由伊蚊(Iridomyrmex detectus)的防收性分泌物中分得的物质。自1958年的Halpem和Schmid确定的环烯醚萜的基本骨架以来,各国学者对该类化合物作了大量深入的研究。环烯醚萜类化合物具有多种生物活性,近来受到极大关注,发展也很迅速。 环烯醚萜类主要分为:环烯醚萜类、裂环环烯醚萜类、3,4-位无取代的环烯醚萜类、聚合环烯醚萜类等等。本人做的是普通类的环烯醚萜类化合物,且以其苷居多,做的比较浅,下面斗胆一谈,各位看官莫要见笑。提取部分 环烯醚萜苷类化合物在醇(甲醇、乙醇)中溶解度较好,部分苷类在水中溶解度也很好。本人在对某植物进行提取的时候,实际上并非针对这类化合物,采用的是60%的乙醇/水,是为了兼顾各类成分。后来在实验过程中发现,60%的乙醇/水条件下,这类化合物的提取率是很高的。 注释:其实如果要针对性分离,可以将提取液简单处理后进行D101大孔柱色谱,对环烯醚萜类化合物进行富集。萃取部分 提取之后,将药液进行浓缩,至无醇味混悬于水中,然后进行萃取。萃取的过程为:等体积的环己烷、乙酸乙酯、正丁醇分别萃取三次,合并各层提取液浓缩得各层浸膏。就目前实验进展情况来看,环烯醚萜苷类化合物主要集中在正丁醇层,水层也有一部分(我目前还没开始这部分工作)。 注释1:萃取的过程,涉及到溶剂的回收,由于这类化合物在高温下不太稳定,所以用旋转蒸发仪进行减压回收溶剂的时候,温度不能过高,我采用的温度是60度(其实60度已经很高了,但是没办法,不设60度,正丁醇回收不了)。 硅胶柱色谱 正丁醇层进行硅胶柱色谱分离,采用氯仿/甲醇梯度洗脱,样品500g,拌样硅胶1500g,柱床硅胶500g,洗脱梯度为50:1→20:1→10:1→5:1→2:1→1:1→0:1。实验的过程中发现,环烯醚萜类化合物,主要集中在氯仿:甲醇=10:1和5:1部分。 注释1:选择硅胶柱色谱,也是人之常情,此处也可选择D101大孔柱色谱,对这类化合物进行富集; 注释2:选择氯仿/甲醇系统,是因为经过小试,此系统对样品分离较好,最重要的,样品在此系统中成点性很好; 注释3:拌样1500g,柱床500g,你没有看错,我没有写错。书上说,拌样:柱床在1:1-1:10甚至1:20或者1:50,而我这里却是3:1。我可以很负责任地告诉你,没有必要按书上的说法,柱床500g足够了,分离效果一点也不差。以前做另外一个植物,拌样用了3000g,柱床才600g,分离效果也不差,一点问题都没有; 注释4:梯度的选择,建议6-8个。梯度太少,各流分成分可能过于复杂;梯度太多,后续分离麻烦。这种大型硅胶柱色谱,属于平常所说的“粗分”,不宜太多,不宜太少,不然就是给自己找麻烦。http://ng1.17img.cn/bbsfiles/images/2010/10/201010061143_249374_1745326_3.jpgODS柱色谱 包括开放型ODS柱色谱和中低压型ODS柱色谱,其实原理一样,只是规模大小不同而已。 我将5:1洗脱的样品进行中低压ODS柱色谱,采用甲醇/水梯度洗脱,水→10%甲醇/水→20%甲醇/水→30%甲醇/水→50%甲醇/水→甲醇,实验结果表明,ODS柱色谱对此类化合物具有良好的分离效果。 注释1:环烯醚萜苷类化合物一般极性较大,一般集中在10%甲醇/水、20%甲醇/水、30%甲醇/水部分; 注释2:水洗下来的,一般为糖苷类化合物,我从此流分中分离得到几个糖类化合物(题外话); 注释3:ODS柱色谱可以多次进行,反复纯化,利用ODS柱色谱可以得到部分单体化合物;http://ng1.17img.cn/bbsfiles/images/2010/10/201010061144_249375_1745326_3.jpg(注:此图为未合并相同流分前的点板情况)制备液相(反相) 从ODS柱色谱上洗脱的样品,经过分析,可以考虑进行制备液相,半制备液相等等。 事实上,一般而言,PHPLC也是获得环烯醚萜类单体化合物最重要的手段之一。 流动相可采用甲醇/水,若峰形不好,可加入少量乙酸改善(这一招屡试不爽)。 波长的选择,可以使用230nm、240nm都可以(我们有PAD检测器,验证过)。其它一些化合物,由于连上芥子酰基,对羟基香豆酰基,还可以选择320nm的吸收。 色谱柱一般是C18的居多(目前未使用过其它柱子),品牌好像都还行(我们主要使用YMC)。凝胶其它填料 由于分离原理的缘故,使用凝胶对环烯醚萜这一类化合物(分子量相差不大,且结构极为类似)进行分离,前景似乎并不明朗。但是,用凝胶将这一类化合物与其他类型的化合物分离,效果还是很不错的。 其它如大孔树脂,前面提过,用来富集是个不错的选择;聚酰胺,我没有使用过,不过从其分离原理来看,对这类化合物不会很敏感。显色剂的选择 部分环烯醚萜类在254nm紫外下有暗斑,这个很实用; 最常用的是浓硫酸-香草醛显色剂:母核上有羟基取代的显蓝色;母核上无羟基取代显紫红色(非绝对); 其它显色剂如硫酸乙醇、碘等等都可以,我个人偏爱浓-香显色剂。结构测定与解析(简) 环烯醚萜类化合物进行NMR测试,首先氘代甲醇,这个没有任何疑问。 我在一些文献中,也看到一些特例,比如使用重水、氘代DMSO,这些基本可以忽略。 关于结构解析部分,此处不再赘述。







 我要推广仪器
我要推广仪器
 下载APP
下载APP