八氯二丙醚的检测方法都有哪些?我只知道一个,SN/T 1774-2006进出口茶叶中八氯二丙醚残留量检测方法--[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,还有其它的么?谢谢
急需SN/T 1774 《进出口茶叶中八氯二丙醚残留量检测方法》。哪位有?不胜感激!
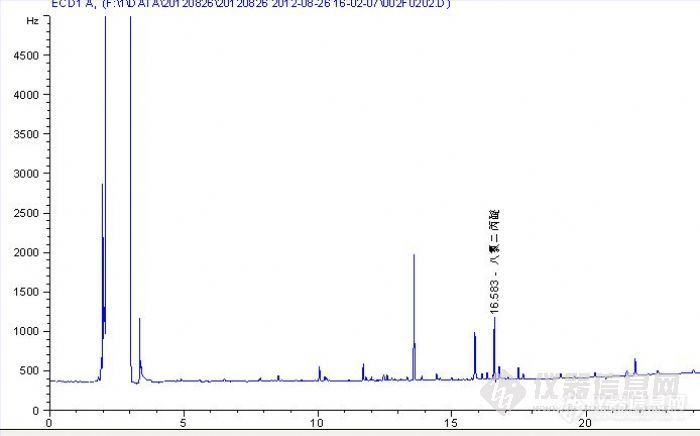
上个月新配了八氯二丙醚的对照品,浓度为0.005ug/ml,图谱如下图一,两个星期后,再次进样,图谱如下图二,请问是不是说明八氯二丙醚降解了呢?求指点,谢谢!http://ng1.17img.cn/bbsfiles/images/2012/09/201209072046_389319_2484453_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/09/201209072046_389320_2484453_3.jpg
大家好!俺是个新手,在此有个问题想诸位请教一下! 请问八氯二丙醚的出峰温度段在哪?以及有机磷和菊酯的出峰段?能否明显区分开来,谢谢!请把分析条件付上,谢谢
请教茶叶中八氯二丙醚残留的GC—ECD测定方法,用什么柱子,大致的前处理方法。
甘油,盐酸,二氯丙醇,氯丙二醇的混合物,要提纯二氯丙醇,各位大人有什么好办法吗?二氯丙醇当然是含量比较多,有试过用乙酸乙酯和无水乙醚来萃取,效果不佳啊?
做蔬菜样品时,先加的水和丙酮,加NaCl分层后,上层应该是丙酮吧?但为什么开始加的丙酮比水多很多,分层后丙酮却比水相少很多呢?而且色素全在丙酮里。 分出来的水相再加二氯甲烷提,分层后下层是二氯甲烷吗?怎么也有人说加了NaCl使二氯甲烷在水的上层呢?
师傅们帮帮我吧,两天了还是找不到二氯丙烷。检测了1千PPM,50PPM,2PPM的二氯丙烷,丙酮做溶剂但是都看不出有明显变化的峰,有两个出峰时间都是1.2跟1.4定性时分别选中这两个峰离子碎片都是43跟58。后来干脆用丙酮直接进样结果也是这两个峰。然后又用二氯丙烷直接进样出峰时间也是1.2跟1.4。不过选中这两个峰时,离子碎片显示是62跟76。应该两个物质重叠了?会不会我设置有问题呢?上图帮忙看下http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_667241_3076231_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016030210524066_01_3076231_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016030210524786_01_3076231_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016030210524830_01_3076231_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016030210524779_01_3076231_3.jpg
双(2-氯异丙基)醚和双(1-氯异丙基)醚是一个物质吗?因为双(1-氯异丙基)醚没有写CAS号所以查不到,也搜不到它[img]https://ng1.17img.cn/bbsfiles/images/2020/03/202003091131393926_7679_3974884_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2020/03/202003091131397330_1037_3974884_3.png[/img]
朋友们谁有苯甲酸甲酯、对甲苯磺酸、TEBAC、甲醇钠、4-二甲胺基吡啶、苯甲酰氯、乙硫醇、巴豆醛、乙酰乙酸甲酯、丙酰氯、DCP的国标、行标或企业标准啊?帮忙找一下啊,谢谢!
各位老师,现在实验室在做氯丙醇脂肪酸酯,参照标准[font=Verdana, Arial][color=#333333][back=#f4f1e2]GB 5009.191-2016 食品安全国家标准 食品中氯丙醇及其脂肪酸酯含量的测定。其中衍生剂七氯丁酰基咪唑需要用气密针吸取,但操作过程中衍生剂容易变成白色固体,用气密针吸不起来,所以改成[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]吸,但好像衍生效果不好。请教各位是不能用[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]吸吗?为什么标准中规定要用气密针?万分感谢[img]https://simg.instrument.com.cn/bbs/images/default/em09512.gif[/img]。[/back][/color][/font]
在做土VOC时,数据分析处理时发现丙酮,二氯甲烷和二硫化碳值都很大,主要是丙酮和二氯甲烷,有几百万那么大。做土svoc和voc时,是分房间处理的,就算交叉污染也不应该值这么大吧……纯水高我可以理解,没加内标和替代,但实验室空白和样品值那么大,我就无法理解。离子源液洗过了,再做一遍值有几万了,但还是太大。二氯甲烷可能跟我这儿的纯水机有关,我这儿用的不是屈臣氏,但丙酮那么大我就一点头绪都没有了……请问问有没有做土水气voc的大神解答一下,谢谢??![img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/07/201907220838277197_6211_3862253_3.png[/img]
求分离一氯丙酮、1,3-二氯丙酮、1,1-二氯丙酮、1,1,3-三氯丙酮、1,1,1-三氯丙酮、1,1,1,3-四氯丙酮的GC分析方法?
最近实验室要建丙二醇丁醚残留的方法,要考虑实验室设备配置问题,请问大家实验室都是用的什么色谱柱?
八溴醚,八溴二苯醚,八溴联苯醚是一样的阻燃剂吗?同时求购八溴联苯醚和五溴联苯醚。[em09508]

3-氯-1,2-丙二醇是丙三醇上的一个羟基被氯原子取代后的化合物,是食品中的污染物,具有致癌性。目前酱油中3-氯-1,2-丙二醇主要来源于水解植物蛋白的副产物,其形成因素主要有蛋白原料的残留脂肪、高浓度的氯离子、大量过剩的酸、高回流温度以及较长的反应时间。2762中规定了酱油里3-氯-1,2-丙二醇的限量为0.4mg/kg。 本次3-氯-1,2-丙二醇(3-MCPD)的检测方法采用GB 5009.191-2016[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱-同位素内标法,样品中加入内标D[sub]5[/sub]-3-氯-1,2-丙二醇(D[sub]5[/sub]-3-MCPD),经硅藻土SPE柱净化,与标准系列溶液一起以七氟丁酰基咪唑衍生后(反应方程式如图1),增强其挥发性,再用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱仪测定,内标法定量。试验过程中遇到一些问题,希望与大家分享交流。[align=center][img=,611,268]http://ng1.17img.cn/bbsfiles/images/2017/08/201708201537_01_2485497_3.png[/img][/align][align=center]图1 衍生反应方程式(上图为3-MCPD,下图为D[sub]5[/sub]-3-MCPD)[/align]1、 色谱条件的确定 由于刚接触[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用法,具体的操作流程还不太熟悉。刚开始未进行全扫描采集定性,直接采用SIM扫描,并且还设置了溶剂延迟,衍生后结果均未找到衍生峰。后期用衍生后标准溶液全扫描后发现衍生峰竟在溶剂延迟前出峰,根本就没有采集到。为避免此类事故的发生,由此制定了以下流程。 本法采用HP-5 MS弱极性毛细管柱,采用梯度升温的方法,先对标准溶液的HFBI衍生物进行全扫描质谱采集,将得到的总离子流图(TIC图)提取色谱图,进行定性,找到目标物3-MCPD和内标D[sub]5[/sub]-3-MCPD衍生产物的峰位置。由此可设定溶剂延迟时间以保护灯丝寿命,目标物出峰后的程序升温过程即冲洗柱子将残留在柱子中杂质冲出,这段时间可不进入质谱,以减少对离子源的污染。 然后采用选择离子扫描(SIM)采集,3-MCPD衍生物特征离子:m/z 253(定量离子)、m/z 275、289、291(定性离子);D[sub]5[/sub]-3-MCPD衍生物特征离子:m/z 257(定量离子)、m/z 278、294、296(定性离子)。由得到的峰面积内标法定量,离子丰度比定性。2、 衍生试剂及衍生条件的确定 常用的衍生试剂有七氟丁酰基咪唑(HFBI)和七氟丁酸酐(HFBA),但HFBA产生的副产物七氟丁酸可能会降解氯丙醇衍生物,因此选择HFBA。 前期试验中两次衍生的结果均未找到目标物的衍生峰,找了好久的原因,包括比对定量离子对和定性离子对、探究反应机理、排查标准物质、定容试剂以及衍生试剂,最终发现衍生试剂HFBI和HFBA混淆,误用了HFBA,导致未找到衍生物峰。本实验室两种衍生试剂都有,试验过程中因疏忽大意,将两者混淆,着实给我们一个教训,实验前一定做好准备工作;另外若发现试验出现问题,寻求原因时可从源头开始,从标准品和试剂着手排查原因。3、 线性范围的选择 在日常检验中,经常会有检出量特别大甚至超标的样品,很有可能超出了线性范围,这样标曲就没有意义了。遇到这种情况可有两种解决方案。一是将标曲线性范围扩大,多衍生几个标曲点,确保样品中被检测物含量在线性范围内;二是减小称样量,若称样量少不具有均一性和代表性的话,还可在检测过程中做稀释处理,使样品含量在线性范围内。4、 硅藻土SPE柱 硅藻土具有很好的吸水性,可降低3-氯-1,2-丙二醇在水相中的溶解度。我们选用成品硅藻土SPE柱(Agilent Chem Elut)与自制硅藻土柱(脱脂棉-硅藻土-无水硫酸钠)进行比对。发现回收率相差不太,均在95%~110%之间。从净化效果来看,差异也不大,从节约成本方面综合分析,还是选用自制硅藻土柱。 选用正己烷作为淋洗溶剂,可去除一些脂溶性杂质。最后用乙酸乙酯作为洗脱液将3-氯-1,2-丙二醇全部洗脱。5、 内标法数据分析 将采集的数据定性后确定保留时间,设置内标化合物及其浓度,建立标准曲线的级别和浓度,内标法定量。 在此过程中,若样品中目标物有共流出现象,定量离子有干扰,积分不准确的情况下,①可以重新优化前处理方法-SPE净化;②若其他特征离子没有干扰且灵敏度足够高的情况下,亦可将此离子作为定量离子,重新分析,离子丰度比需在规定范围内。 通过这次试验,让我明白了每一个项目的方法开发、每一批样品的检测都应做好充分准备,包括标准溶液的配制和比对,所用试剂的种类、浓度和有效期,前处理步骤的先后顺序、时间长短以及关键点,检测器、色谱柱、流动相的选择,数据分析的全面性(保留时间、定性离子、定量离子、NIST库);提前查阅文献和资料熟知试验过程及每个步骤的作用和原理,不得有半点马虎,否则做一些无用功,即浪费资源又浪费时间。希望通过这次3-氯-1,2-丙二醇检测中的亲身体会,与大家分享,也告诫一下自己,不断努力,不断提高自身的技术水平。
国标GB 5009.191-2016 做3-氯-1,2-丙二醇用七氟丁酰基咪唑进行衍生化,但衍生剂很容易变质,用的时候不方便。若是能用正己烷或其它溶剂稀释后可能会好用一些,有人试过吗?
酱油中3-氯1,2-丙二醇的测定方法??有没有版友做过这个项目呀?不用MS 只用GC作。用七氟丁酰基咪唑衍生。
在测定挥发性有机物时给它定性时 二氯丙烷的相似度检索出来的相似度为98的定量离子和国标上的参考离子的完全不一样 那它是不是二氯丙烷 国标上的参考离子为 63 41 112 ms中是63 62 76
求大佬指导,怎样用GC-MS测试试剂乙硫醚,二甲二硫醚,2-甲基-2-丙硫醇,1-丙硫醇,有没有参考文献?
国标GB 5009.191-2016 做3-氯-1,2-丙二醇用七氟丁酰基咪唑进行衍生化,但衍生剂很容易变质,用的时候不方便。若是能用正己烷或其它溶剂稀释后可能会好用一些,有人试过吗?
替人问下:大家知道哪里有以下优级纯,分析纯的试剂卖:五溴二苯醚和八溴二苯醚
工业二氯丙烷分析方法谢谢
卤代酚卤代酚是含酚的卤代化合物,对革兰氏阳性菌有强杀菌作用,用在化妆品中的卤代酚有六氯酚 等多种化合物。这类化合物通常是光敏物质。我国化妆品卫生标准规定为限用物质,限用量见表2-3-17。表 2-3-17 化妆品卫生标准中卤代酚的限用量品名 序号 最大使用量(%) 溴氯双酚 4-4 0.1 双氯酚 4-7 0.2 2,4-二氯二甲苯酚 4-8 0.1 三氯生 4-21 0.3 六氯酚 4-24 0.1 4-溴邻甲苯酚 4-31 0.3 苄氯酚 4-42 0.2 4-氯2-甲苯酚 4-55 0.2 4-氯3,5-二甲苯酚 4-56 0.2 * 指化妆品卫生标准(GB7916-87)中的序号,4-42即表4的序号42(一)薄层色谱法(TLC)1 适用范围本方法适用于化妆品中六氯酚,双二氯酚硫醚、二氯酚和三溴水杨酞替苯胺的定性。2 原理样品经预处理后,样液中的卤代酚用的薄层色谱法进行分离、呈色,然后与标准斑点比较,进行定性。3 试剂3.1 乙醇:分析纯。3.2 己烷:分析纯。 3.3 丙酮:分析纯。3.4 无水硫酸钠:分析纯。3. 5硫酸(lmol/L)。3.6 六氯酚标准溶液(1):准确称取用苯重结晶的六氯酚50.0mg,加丙酮溶解后移入50ml容量瓶中并定容至刻度,避光保存。此溶液1ml含1.0mg六氯酚。3.7 双二氯酚硫醚(2):准确称取用苯重结晶的双二氯酚硫醚50.omg,用丙酮溶解,移入50ml容量瓶中并定容至刻度,避光保存。此溶液lml含1.0mg二氯酚硫醚。3.8双氯酚标准溶液(3):准确称取用甲苯重结晶的双氯酚50.0mg,用丙酮溶解,移入50ml容量瓶中,定容至刻度。此溶液1.0ml含1.0mg二氯酚,避光保存。3.9三溴水杨酞替苯胺(4):准确称取用丙酮重结晶的三溴水杨酞替苯胺50.0mg,用丙酮溶解,移入50ml容量瓶中并定容至刻度。此溶液1.0ml含1.0mg三溴水杨酞替苯胺,避光保存。3.10 乙醇一己烷(1 9)。3.ll离子交换纤维素(5):将DEAE(二乙基氨基乙醇)纤维素,(交换量约0.9meg/g),浸泡于50倍量的0.lmol/L的盐酸中,用玻璃漏斗过滤,用20倍量的丙酮,30倍量的0.lmo1/L氢氧化钠溶液淋洗至OH-型后,用水洗成中性,再用20倍量的丙酮淋洗,弃去丙酮。空气中干燥。保存在乙醇十己烷(l+9)溶液中。3.12 硅胶:薄层用硅胶中加有荧光剂。3.13碱性氧化铝。3.14展开剂:石油醚 冰乙酸(89 12)3.15显色剂。3.15.1 浓氨水。3.15.2 2%4-氨基安替比林溶液:称取2g4-氨基安替比林用乙醇溶解稀释至100ml。3.15.3 8%铁氰化钾溶液(K3[Fe(CN)6])。3.15.4 2%三氯化铁溶液(FeCl36H20):称取2g三氯化铁用乙醇溶解稀释至100ml。3.15.5 2%铁氰化钾溶液。3.16 盐酸 丙酮溶液:9.5ml盐酸加丙酮至100ml(临用前配制)。4 仪器 4.1 层析柱:、内径10mm、高200mm的具塞玻璃管的下端熔接玻璃过滤器或塞有玻璃棉,4.2 紫外灯,具有8W功率,254nm波长。4.3离子交换柱(6):将离子交换纤维素用乙醇 已烷(3.11)配成混悬液,:用湿式填充法缓慢倾入层析柱中,以防止产生气泡,填充高度80mm。5 分析步骤5.1样品预处理(7)(8)称取含卤化酚0.5mg的样品(扑粉,除臭砂芯、香波约10g,膏霜约0.5g),置于100ml玻璃瓶中,连接好回流冷凝器:加50ml乙醇 已烷(1 9)溶液,2ml 1mol/L硫酸,于水浴上加热3min,冷却后用3号玻璃砂芯漏斗过滤,用乙醇 己烷(1 9)溶液5ml洗沉淀,滤液移入分液漏斗中静置分层。取己烷层用10ml水洗涤,无水硫酸钠脱水后以0.5ml/min的流速注入离子交换柱(10)。用50ml己烷洗涤。去除油脂等干扰物质,弃去淋洗液,依次用10ml丙酮、2ml丙酮 盐酸溶液(3.16),20ml丙酮洗脱(11)。溶出液在水溶上加热蒸去有机溶媒,加5ml乙醇,加热使盐酸挥发,重复此操作2次。残渣加2.0ml丙酮溶解,作为样品待测溶液。5.2 制备薄层板5.2.1硅胶薄层板:硅胶30g,加水约65ml,搅拌均匀,涂布成厚度0.25~0.3mm的薄层板,105~l10℃干燥30min,置干燥器中保存。 5.2.2含硝酸银的氧化铝薄层板:0.12g AgNO3,加少量水溶解,加30ml乙醇、20g氧化铝,调成浆状物,涂布厚度为0.25~O.3mm的薄层板,空气中干燥、于干燥器中避光保存。5.3 点样距薄层板底边2cm处将5~20μl待测溶液从左到右点样(12),两点间隔约1cm,薄员板.的右边点2μl标准溶液,空气中干燥。5.4 展开取适量展开剂(3.14)倾人展开槽中,将薄层板放入展开剂中,待溶剂上升约10cm,取出薄层板,空气中干燥。5.5显色(13)在薄层板上顺序喷雾显色剂3.15.1~3.15.3或3.15.4~3.15.5,六氯酚在显色剂3.15.1~3.15.3中为红色,在3.15.4~3.15.5中为蓝色斑点。二氯酚硫醚和三溴水杨酞替苯胺在3.15.1~3.15.5中为紫色,在3.15.4~3.15.5中呈现蓝色斑点。用加荧光剂的硅胶薄层板测定时,各种卤代酚在紫外线照射下,在各自的Rf 值位置上以荧光为背景呈现出暗黑色的斑点。
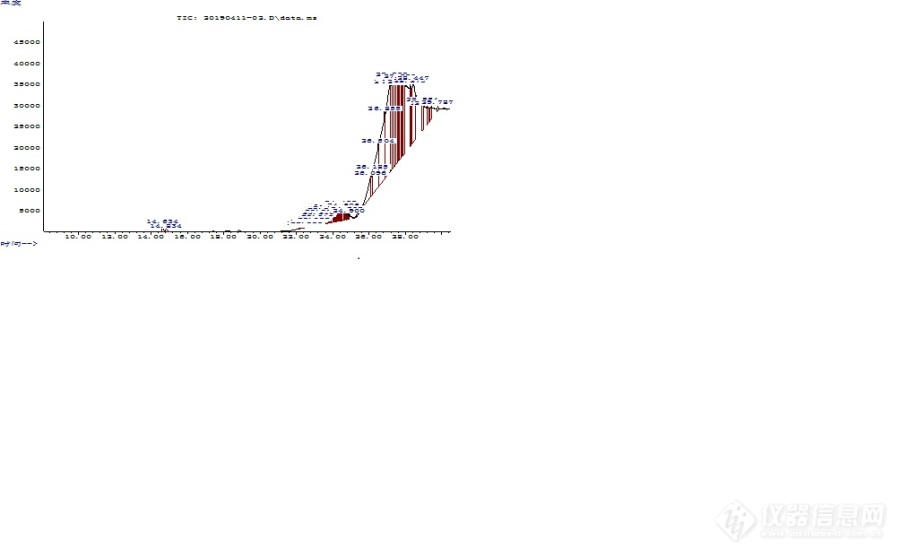
最近在开展食品中3-氯-1,2-丙二醇方法摸索,在对标样进行衍生,并作标准曲线的时候发现有如下问题,请各位大神不吝指教。1、选取5个点作为3-氯-1,2-丙二醇标准样品,质量分别为0.0286ug、0.143ug、0.286ug、0.572ug、1.144ug,在作标准曲线时,在每个标样中分别加入等量的D5-3-氯-1,2-丙二醇,其质量为0.222ug,上述标样中均加入正己烷定容至2mL,然后按标准要求加入40uL的七氟丁酰基咪唑进行衍生反应,后续的步骤严格按标准要求执行;2、衍生结束后,上[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]进行测定,这时候问题出现了,按照上述的内标加入量,D5-3-氯-1,2-丙二醇在各标样的中的量应该是相同的,按理论来说,其在谱图中的峰面积也应该是大致相同的,最起码不会有太大的差别,但实验结果却是内标的峰面积会随着标样浓度的增加而不断的增大,其具体的数值为38845、86170、185376、412975、887632;反观标样3-氯-1,2-丙二醇的峰面积,其实测值与标样浓度值对应增长,成线性关系,具体测定值分别为26801、64667、138047、300758、640761。如果按照外标作标准曲线的话,线性是没问题的,但如果按内标作曲线的话,作出来的曲线压根就没法使用,可以说完全不成线性,这就是我请请教各位同行的问题,你们在用内标作这个项目的时候也会遇到这个问题吗?如果没有,那么你们的内标面积都能保持一致吗?为了各直观的说明这个问题,我把我做的5个标准点谱图按浓度从低到高一并附上以作参考,其中前面的峰为内标,后面的峰为3-氯-1,2-丙二醇。[img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151752487962_2765_1640909_3.jpg!w690x425.jpg[/img][img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151752589614_3231_1640909_3.jpg!w690x425.jpg[/img][img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151753062214_3758_1640909_3.jpg!w690x425.jpg[/img][img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151753121534_1266_1640909_3.jpg!w690x425.jpg[/img][img=,690,425]https://ng1.17img.cn/bbsfiles/images/2019/04/201904151753188554_5523_1640909_3.jpg!w690x425.jpg[/img]
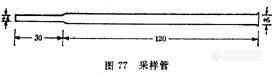
空气中二氯丙醇的测定方法 变色酸比色法 1 原理二氯丙醇水解后,经高碘酸氧化生成甲醛。甲醛与变色酸作用生成紫色化合物,比色定量。2 仪器2.1 采样管(图77)。2.2 抽气机。2.3 流量计,0~1L/min。2.4 具塞比色管,25ml,10ml。2.5 分光光度计。3 试剂3.1 硅胶:40~60目硅胶,用混合酸(硫酸与硝酸等体积混合)在沸水浴上回流加热处理2h。水洗至中性,在110℃干燥4h,然后在200℃活化4h,贮于干燥器内备用。[img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201424_52378_1625938_3.jpg[/img]3.2 碳酸钠(Na2CO3)溶液,10g/L。3.3 高碘酸钾溶液,15g/L:称取1.5g高碘酸钾,溶于100ml3+2硫酸中。3.4 亚硫酸钠溶液,100g/L。3.5 硫酸,?20=1.84g/ml。3.6 变色酸溶液,20g/L。临用前配制。3.7 标准溶液:于25ml量瓶中加入约10ml碳酸钠溶液(3.2),准确称量,滴入2滴二氯丙醇,再准确称量,两次称量之差即二氯丙醇的质量,加碳酸钠溶液(3.2)至刻度,充分混合,计算1ml溶液中二氯丙醇的含量。临用前用碳酸钠溶液(3.2)稀释成50?g/ml二氯丙醇标准溶液。4 采样于采样管中先投入一个玻璃珠,使其恰好堵在采样管的下端狭窄部位,然后装入3g硅胶,以0.5L/min的速度抽取5L空气(采样管始终保持垂直位置)。5 分析步骤5.1 对照试验:同采样,于采样管中装入硅胶带至现场,但不抽取空气,照样品分析。5.2 样品处理:将采样管中的硅胶移入25ml比色管中,加10ml碳酸钠溶液(3.2),盖上磨口塞(不要塞严),放在沸水浴中加热90min,放冷,取2ml上清液于10ml比色管中。5.3 标准曲线的绘制:按表71配制标准管。向标准管中各加入0.2ml高碘酸钾溶液(3.2),混匀,放置30min,加入0.2ml亚硫酸钠溶液(3.4),振摇(此时溶液应为无色,如残有黄色可再补加一滴亚硫酸钠溶液),沿管壁徐徐加入3ml硫酸(3.5)及0.6ml变色酸溶液(3.6),混匀,放在沸水浴中加热20min,放冷,加水稀释至10ml,混匀,于波长570nm下比色。以二氯丙醇含量对吸光度作图,绘制标准曲线。5.4 测定:样品管操作同标准管,以现场对照管调仪器零点比色。由标准曲线上查出二氯丙醇的含量。6 计算表71 二氯丙醇标准管的配制[img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201425_52379_1625938_3.jpg[/img]X=5C/V0式中:X——空气中二氯丙醇的浓度,mg/m3;C——所取样品溶液中二氯丙醇含量,微克;V0——标准状况下的样品体积,L。7 说明7.1 本法检测限为1微克/2ml。二氯丙醇浓度为1.5、10、20微克/2ml时,变异系数分别为1.4%、2.6%、2.3%。7.2 采样速度为0.5~1.0L/min时,硅胶对二氯丙醇的采样效率接近100%。
急切求助用气相色谱检测调味品中3-氯,1,2-丙二醇的检测方法,我用国标方法,过柱后的洗脱液好像有水分,衍生很难进行下去,有什么好的净化方法吗?有合适商品柱吗?
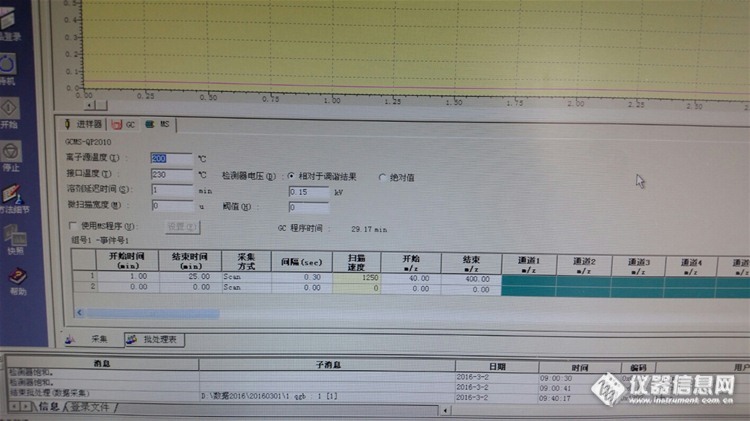
仪器是QP GCMS-2010 SE 现在公司要求检测产品中的二氯丙烷。这个物质我没有做过标线。该怎么找条件,怎么计算大概出峰时间,怎么设置GC跟MS的条件呢?之前的别的物质标线是工程师帮我弄的。现在碰到这情况,我是一无所知了。各位师傅们请多多指教!谢谢你们了!
最近做3-氯-1,2丙二醇这个项目,购买一小瓶97%七氟丁酰咪唑,只有大约1ml,感觉直接加纯品衍生化太浪费,想用什么溶剂制成溶液再使用,请问这样可不可以?如果这样用什么溶剂溶解比较合适?http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif
现在要做一项溶残检测,同时含二氯甲烷、异丙醇和乙腈,在DB-624(30m*320um*1.8um)柱上异丙醇和乙腈完全重叠,在HP-INNOWax(30m*320um*0.25um)柱上异丙醇和二氯甲烷的分离度很低(小于1.0)。现在是顶空进样方式,分流比5:1,调节了流速等其他参数,都不能达到理想的分离效果,请教各位同仁有没有能较好分离这三种溶剂的色谱条件,多谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP