如题:氨基酸态氮含量等同于氨基酸含量吗?
总的氨基酸含量 游离氨基酸含量 氨基氮含量 总氮含量的区别?
湖北省食品质量安全监督检验研究院组织的酱油中氨基酸态氮含量的能力验证。有参加的朋友进来交流交流
湖北省食品质量安全监督检验研究院组织的酱油中氨基酸态氮含量的能力验证。有参加的朋友进来交流交流。
GB5009.39-2003中甲醛值法测定样品中氨基酸态氮的含量,甲醛已经不是澄清透明,而是白色糊状的了,可以用吗?用的话对测定结果是不是影响很大,会是什么样的影响呢?
山葡萄酒含有22种氨基酸,其中人体必需的8种氨基酸(指人体自身不能合成的氨基酸)有苏氨酸、缬氨酸、异亮氨酸、亮氨酸、苯丙氨酸、赖氨酸、蛋氨酸、色氨酸等,总量为1350mg/L,占总氨基酸含量的45.6%,是一般葡萄酒的10~20倍;而一般葡萄酒只含有6~7种人体必需的氨基酸。
通常,饲料原料的氨基酸含量是通过离子交换(IEC)或高效液相色谱(HPLC)方法测定。这些方法由于费用极其昂贵,而且所需时间很长,用于饲料厂的配方调整和质量控制,不切实际。也可以通过测定饲料原料的干物质和粗蛋白含量,回归预测氨基酸含量(NRC,1998),但是,这种折中的方法推测的饲料原料的氨基酸含量与其实际值差异较大(丁丽敏等,2002),直接用于生产,会造成较大的损失。在实际生产中,饲料厂在制定饲料配方时,饲料原料中所用的氨基酸含量只好参考数据库中推荐的平均值,但推荐的平均值无法反应特定原料氨基酸含量的变异。因此,饲料研究和生产人员一直在寻找一种快速、廉价的测定饲料原料氨基酸含量的方法。 过去人们认为,近红外(Near-Infrared Reflectance Spectroscopy,简称NIRS)技术不适用于测定饲料原料氨基酸含量,因为原料中氨基酸含量过低。但是,近年来,随着近红外分析技术和仪器的发展,通过近红外技术测定的氨基酸结果与传统的测定方法具有了很好的可比性(张维军等,2000)。本文主要对笔者近期应用NIRS 方法快速测定饲料原料氨基酸含量方面所取得的经验进行介绍,以使NIRS技术更好的服务于国内饲料行业。 1 NIRS技术测定饲料原料氨基酸含量的原理和方法 NIRS技术是依据被检测样品中某一化学成分对近红外区光谱的吸收特性而进行定量测定的一种分析方法。应用NIRS方法测定原料氨基酸含量,首先要建立定标方程。定标方程的建立,不仅需要饲料原料的NIRS光谱值,而且需要利用标准方法测定的原料的氨基酸含量。饲料原料NIRS光谱的扫描和氨基酸含量的化学分析值必须是对同一样品进行的。根据所得的光谱值和化学分析值,运用多元回归计算便可得到相应的定标方程。但仅有很少的实验室能够大量的测定这些数据,这也是NIRS在测定原料氨基酸含量方面发展较慢的原因。 每种饲料原料至少需要50个样品才能建立定标方程,而且所取样品必须具有代表性。增加建立定标方程所用的样品数量,能够提高定标方程的准确性和可靠性。德固赛(Degussa)公司拥有全球最大的饲料氨基酸分析实验室,每年分析的饲料样品超过10,000个,为建立NIRS定标方程奠定了坚实的基础,目前,已经对全球16种常用饲料原料的干物质、粗蛋白质和所有必需氨基酸含量建立了定标方程(表1)。新到货的饲料原料,可以对其取样、制样,然后利用NIRS 分析仪对其样品进行扫描获得NIRS 光谱值,并根据相应的定标方程计算,很快就可以获得饲料原料的氨基酸含量。 表1 已经建立定标方程的饲料原料 类别原料名称N 大麦233 玉米328 黑麦273 谷物及其副产品小麦281 麦麸178 高梁205 米糠181 玉米蛋白粉184 菜籽粕171 豆粕341 油籽类葵花粕107 豌豆110 羽扇豆105 鱼粉307 动物副产品羽毛粉247 肉骨粉468 注:N=建立相应饲料原料定标方程的样本数 2 NIRS与色谱法测定的饲料原料氨基酸含量的一致性 NIRS分析结果的有效性,最终还要看其与色谱法测定值的吻合程度(Fontaine等,2001)。图1~3列出了德固赛(Degussa)公司利用NIRS和色谱方法测定的豆粕、肉骨粉和小麦的蛋氨酸以及赖氨酸含量之间的拟合情况。由图以及相应的统计结果可以看出,NIRS方法对饲料原料的氨基酸含量的预测值非常准确和可靠。因此,在实际生产中,可以利用快速、低成本的NIRS 技术代替耗时、昂贵的色谱法测定饲料原料的氨基酸含量。 3 NIRS 技术在饲料生产中的作用 由于利用NIRS技术测定饲料原料氨基酸含量,不仅费用极其低廉,而且所用时间很短,因此获得饲料原料氨基酸含量测定结果的时间主要决定于对原料进行采样和制样的时间。饲料厂可以通过快速、准确的氨基酸含量测定值,评估来自不同产地和供货商的原料,实施真正的质量管理。 3.1 细分饲料原料 饲料厂可以根据氨基酸的测定值,将到厂的饲料原料进行细分。例如,以下几个因素可以作为细分时考虑的因素:品种,种植方法(例如施肥情况),产地,加工条件。将饲料原料细分,能够促使配合饲料厂更准确的优化配方。为了统计分析不同来源、品种或供应商原料的差别,所采集的样品必须具有代表性。而且,每种来源或供应商的样品至少应该有20个。如表2所示,根据粗蛋白质和氨基酸含量的测定值,饲料原料可以分为不同的质量等级。不同质量的饲料原料,应该以不同的价格购买。 表2 根据养分含量对饲料原料(以豆粕为例)的细分表 养分含量(%)所有样品低等质量中等质量高等质量 N=490N=62N=348N=80 粗蛋白(均值)44.8741.6944.8147.58 变异系数4.70 4.632.723.51 赖氨酸(均值)2.7632.5612.7552.961 变异系数4.643.482.511.69 蛋+胱(均值)1.2771.1891.2761.353 变异系数4.693.952.662.89 苏氨酸(均值)1.7321.6081.7291.840 变异系数6.15 4.35 2.89 3.26 注:N=样品数量 如果配合饲料厂以相同的价格购买所有质量级别的饲料原料,那么饲料厂将会为氨基酸含量较低的饲料原料支付过高的价格。因此,当供应商提供的饲料原料氨基酸含量较低时,饲料厂应该调低支付价格或拒绝接货。 3.2 提高配合饲料质量 NIRS技术还能充分保证配合饲料的质量。如果不能及时检测到的饲料原料的养分变异,即使机械设备和混和技术达到最佳,也会严重影响配合饲料的质量。原料氨基酸含量较大的变异,不可避免的需要增加配合饲料的安全系数,从而增加生产成本。在养殖业中,饲料养分的变异,会导致动物生产性能的降低和畜产品质量变异的增加,尤其是像胸肉率和瘦肉率这些敏感指标。畜产品质量的降低或不整齐,会导致其价格的降低。如果饲料原料到货时以及随后能经常性的检测其氨基酸含量,饲料厂则可以根据检测的结果,对配方进行及时调整,从而保证配合饲料的氨基酸含量。 4 NIRS技术推广中存在的问题与对策 NIRS 技术能够降低饲料生产成本,提高饲料产品质量,但是许多饲料厂没有购买NIRS 分析仪器,有些饲料厂虽然拥有自己的NIRS分析仪器,却没有能力建立可靠的氨基酸定标方程,在一定程度上限制了NIRS 技术的推广。针对以上情况,可以采用一些更为灵活的方法,使饲料厂能够在几乎不增加成本的基础上,获得NIRS技术服务。没有购买NIRS分析仪器的饲料厂,可以将采集的饲料原料样品邮寄到相应单位进行测定。而已经购买了NIRS分析仪器却没有能力建立定标方程的饲料厂则可以与建立了NIRS 饲料原料氨基酸含量定标方程的单位合作,获得定标方程的转移和人员的培训服务。定标方程转移的前提条件是软硬件之间的兼容性。另外,这些饲料厂也可以利用自己的NIRS分析仪对饲料原料进行扫描,然后将获得的NIRS光谱值,通过电子邮件传到拥有NIRS定标方程的单位,进行相应的数据处理,便可以获得饲料原料的氨基酸含量值。通过这些方法,饲料厂可以在几乎不增加成本的基础上,利用NIRS技术,在线检测饲料原料的氨基酸含量。通过在线检测饲料原料氨基酸含量,饲料厂可以更有效的监控原料质量和调整配方,生产出优质、低价的配合饲料产品。 综上所述,NIRS技术测定饲料原料氨基酸含量,具有快速、准确、成本低的特点。因此,饲料厂可以利用NIRS技术对主要的饲料原料氨基酸含量进行在线监测,调整配方和采购策略,降低生产成本,提高产品质量。
应用近红外技术快速测定饲料原料氨基酸含量通常,饲料原料的氨基酸含量是通过离子交换(IEC)或高效液相色谱(HPLC)方法测定。这些方法由于费用极其昂贵,而且所需时间很长,用于饲料厂的配方调整和质量控制,不切实际。也可以通过测定饲料原料的干物质和粗蛋白含量,回归预测氨基酸含量(NRC,1998),但是,这种折中的方法推测的饲料原料的氨基酸含量与其实际值差异较大(丁丽敏等,2002),直接用于生产,会造成较大的损失。在实际生产中,饲料厂在制定饲料配方时,饲料原料中所用的氨基酸含量只好参考数据库中推荐的平均值,但推荐的平均值无法反应特定原料氨基酸含量的变异。因此,饲料研究和生产人员一直在寻找一种快速、廉价的测定饲料原料氨基酸含量的方法。 过去人们认为,近红外(Near-Infrared Reflectance Spectroscopy,简称NIRS)技术不适用于测定饲料原料氨基酸含量,因为原料中氨基酸含量过低。但是,近年来,随着近红外分析技术和仪器的发展,通过近红外技术测定的氨基酸结果与传统的测定方法具有了很好的可比性(张维军等,2000)。本文主要对笔者近期应用NIRS方法快速测定饲料原料氨基酸含量方面所取得的经验进行介绍,以使NIRS技术更好的服务于国内饲料行业。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=69522]应用近红外技术快速测定饲料原料氨基酸含量[/url]
傅里叶近红外能测量出小麦种氨基酸的组成含量吗?这个氨基酸不是和水分淀粉相平行的一个指标,而是小麦当中各种氨基酸的组成,大概有17种,例如:赖氨酸、谷氨酸、丝氨酸等各种氨酸的含量。有哪位坛友知道么?
测定氨基酸含量,除了利用氨基酸与茚三酮的显色反应外,还有什么别的方法?
具体操作,大蒜浸提液在90℃下水浴2h,检测其中一种水溶性非蛋白质氨基酸的含量,该物质的出峰时间是5.14min。在这个图上看不出峰来,是不是要除杂,或者是采取柱前衍生化法。跪求给位色谱前辈指导!
我要用氨基酸分析仪测土壤中游离氨基酸的种类和含量.但不知道如何处理土壤样品 现在非常着急 不只那为好心人可知道如何做 谢谢
复方氨基酸注射液 含量测定 是用液相色谱吗?哪位大侠知道怎么弄的,指导我呀。谢啦! 我13914681818 wcj.yc@163.com
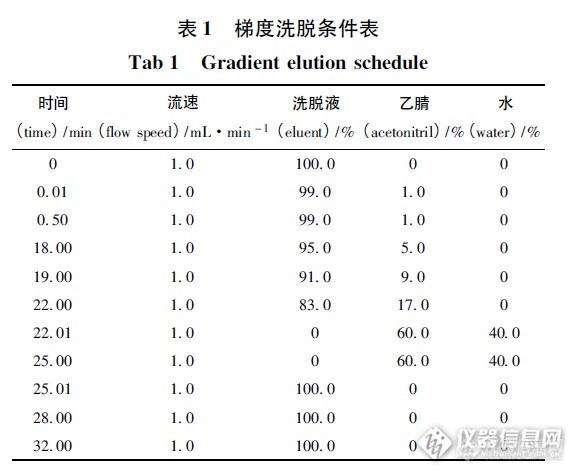
人凝血因子VIII中氨基酸含量测定摘要 目的: 建立用高效液相色谱法测定人凝血因子VIII中氨基酸含量。方法: 采用6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 为衍生剂,与氨基酸柱前衍生后,用Agilent 1200 高效液相色谱仪,AccQ·Tag C18柱( waters 150 mm ×3. 9 mm,4 μm) ,以水Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液和乙腈进行梯度洗脱,检测波长为248 nm,柱温37 ℃,进样量10μL。结果: 各氨基酸在32 min 内测定完毕,回收率为98.7% ~ 101.5%。RSD 均小于1. 5%。结论: 本法分离度好,快速、简便,可作为产品的质量控制方法。关键词: 6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯; 人凝血因子VIII; 甘氨酸; 衍生物; 梯度洗脱; 高效液相色谱法;氨基酸; 含量测定人凝血因子VIII,本品对缺乏人凝血因子礓所致的凝血机能障碍具有纠正作用,主要用于防治甲型血友病和获得性凝血因子Ⅷ缺乏而致的出血症状及这类病人的手术出血治疗。该药物制备过程中使用了氨基酸( 精氨酸、丙氨酸、甘氨酸、组氨酸、盐酸赖氨酸、脯氨酸 等) 做稳定剂,为了保证药品质量和用药安全,应对其中氨基酸的含量进行控制。该法依据过量的6 - 氨基喹啉基- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 在一定条件和氨基酸形成稳定的衍生产物( 柱前衍生) ,用高效液相色谱法测定衍生产物,根据衍生产物的含量计算人凝血因子中各氨基酸的含量。1 仪器和试药1200 高效液相色谱系统( 美国Agilent 公司) ,配置低压四元梯度泵、1314B 紫外吸收检测器、自动进样器、柱温箱、Chemistations 化学工作站; Sartorius CP225D 电子微量天平( 德国Sartorius 公司) ; SartoriusPB - 21 型pH 计( 德国Sartorius 公司) ; LDZ5 -2 低速自动平衡离心机( 上海医用离心机厂) 等。各标准品均来自于中国食品药品检定研究院2 色谱条件及系统适用性试验色谱柱: Waters AccQ·Tag C18色谱柱( 3. 9 mm ×150 mm) ; 流动相: 水为溶剂D,Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液( A) - 乙腈( B) - 水( D) ,柱温:37 ℃; 检测波长: 248 nm。精密量取对照品溶液与供试品溶液10 μL,分别注入液相色谱仪,记录色谱图32 min。梯度洗脱条件表见表1。http://ng1.17img.cn/bbsfiles/images/2015/08/201508311802_563771_1637386_3.png
用氨基酸自动分析仪测定氨基酸含量,配制不同pH的柠檬酸钠缓冲液的作用是什么?是淋洗不同的氨基酸?如果用732阳离子树脂吸附氨基酸,用氨水来淋洗,测定氨基酸总量,原理上可行吗?谢谢!
国标上用氨基酸测定仪来测定氨基酸含量,但我们没有这个仪器,现在想用液相色谱仪来测,好像要用柱前衍生化方法来测,苦于没有相关含测的资料,哪们同仁有的话请在这里共享.谢了.
《化妆品中氨基酸含量的测定》QBT 2409-1998[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=155337]《化妆品中氨基酸含量的测定》QBT 2409-1998[/url]
高效液相色谱测氨基酸含量(SIGMA 混合标样的问题)/17种氨基酸出峰顺序?
我最近在做氨基酸含量测定.用的是PICT衍生法.可是做的标准品(甘氨酸.谷氨酸,ASP都有好几个峰.根本分不出标准品的峰..查了不少文献都是这么做的.哎,真不知道怎么办啊.哪位做过这方面的高手帮帮忙啊.我用的是15厘米C18柱,PDA检测器,
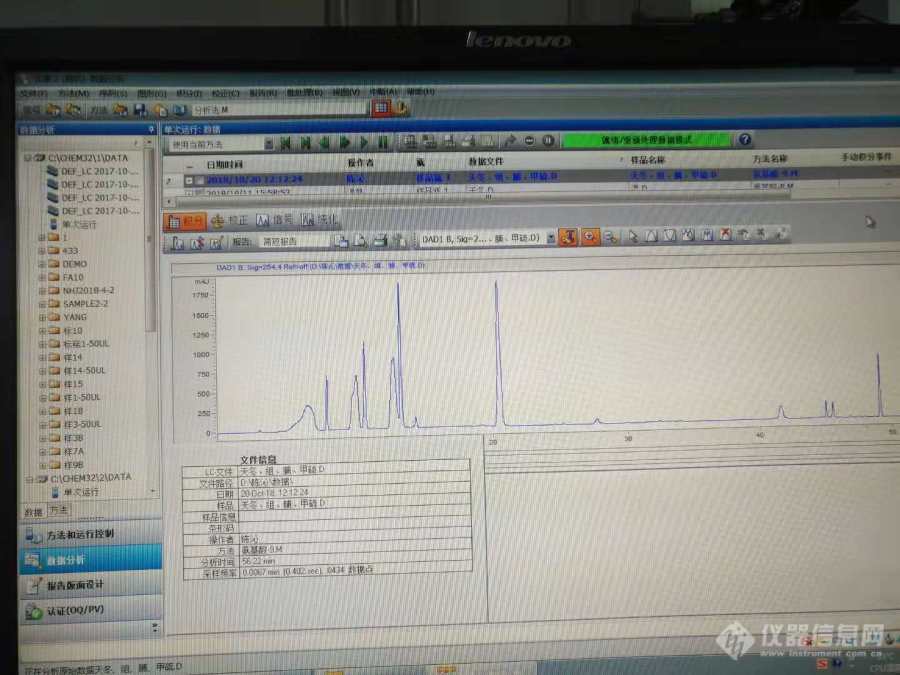
我是参考一篇PITC柱前衍生法测黄酒中氨基酸含量的文献,方法、色谱柱都和文献一样,但是为什么我跑出来的色谱峰有很多都是一种氨基酸对应两个色谱峰。我每次衍生剂都是现配现用的,我换过一根色谱柱,结果还是一样,我不知道问题出在哪里。请大家帮忙分析分析原因,十分感谢。如图1是我跑的四种氨基酸的标品,前面两种氨基酸都是分别对应两个色谱峰。后面两个氨基酸分别是单独的一个峰。图2是我跑的17种氨基酸混合标品.后面两张图是我参考的文献里面的具体洗脱方法.请各位大神给予指点.[img=,554,296]https://ng1.17img.cn/bbsfiles/images/2018/10/201810240941270206_3458_3363167_3.png!w554x296.jpg[/img][img=17种氨基酸混合标品,554,296]https://ng1.17img.cn/bbsfiles/images/2018/10/201810240936371461_9895_3363167_3.png!w554x296.jpg[/img][img=,428,311]https://ng1.17img.cn/bbsfiles/images/2018/10/201810240940035288_3112_3363167_3.jpg!w428x311.jpg[/img][img=参考的文献具体洗脱程序,435,294]https://ng1.17img.cn/bbsfiles/images/2018/10/201810240936544035_7123_3363167_3.jpg!w435x294.jpg[/img]
最近要做阿胶中氨基酸的含量测定,2010cp的方法如下:取本品0.25g,精密称定,置25ml量瓶中,加0.1mol/L盐酸溶液20ml,超声处理(功率500W,频率40kHz)30分钟,放冷,加0.1mol/L盐酸溶液至刻度,摇匀。精密量取上述溶液2ml,置5ml安瓿中,加入等体积的盐酸,150℃水解1小时,放冷,移入蒸发皿中,用10ml水分次洗涤,洗液并入蒸发皿,蒸干,残渣加0.1mol/L盐酸溶液溶解,转移至25ml量瓶中,加0.1mol/L盐酸溶液稀释至刻度,摇匀,即得。前处理方法比较繁琐,安瓿每次都要封口,而且挺危险的,150℃高温水解一般只能用烘箱控温了,大家有什么好建议吗?谢谢!
复方氨基酸注射液采用丁基胶塞密封,胶塞中的硫磺可能会浸出到注射液中,需要对药液中的硫磺含量进行测定。但是,方法学建立过程中,进行药液的加标回收率测定,发现其不合格,并且随氨基酸浓度升高,硫磺回收率降低。求助可能的原因,有哪位大神做过相关的实验吗?请不吝赐教,大谢
哪 位朋友有氨基酸注射液含量的液相检测方法,传过来学习一下。谢谢!
我最近在做氨基酸含量测定.用的是PICT衍生法.可是做的标准品(甘氨酸.谷氨酸,ASP都有好几个峰.根本分不出标准品的峰..查了不少文献都是这么做的.哎,真不知道怎么办啊.哪位做过这方面的高手帮帮忙啊.我用的是15厘米C18柱,PDA检测器,

O(∩_∩)O哈哈~ 运用近红外测定棉籽粕的17种氨基酸的含量,http://ng1.17img.cn/bbsfiles/images/2011/12/201112092223_337051_1639754_3.jpg文献上近红外若能准确的分析原料中的氨基酸含量,得省多少money(我们用的氨基酸分析试剂好贵)? 省多少time(我们一般最少是2d才能出结果)?
哪位大侠以前做过测定游离氨基酸含量啊,我现在在弄这个,搞得我蛮郁闷的原来的25mm的长柱子用1mL/min,做得效果不行,好几个峰连在一起改成短柱,用0.3mL/min的时候,在不调节A相(0.1mol/l的醋酸钠)的pH情况下,能把峰做得比较开(但还不是很好);但调了A相的pH(6.5)后反而不好了,峰都挤在一起我进样量3微升,梯度是标准的梯度(国标)哪位大侠知道的麻烦发个话,小弟做得好郁闷啊
[em53] 请问如何测定低含量氨基酸methyliminodiacetic acid or ethylenediamine diacetate 在高浓度盐溶液中。我想选用阴[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url],选择Na2CO3 and NaOH 配置的缓冲液作流动相不知能否有效。我以前没用过,很头疼[em06]!现谢谢了,各位高手!
请问各位老师,我用岛津高效液相色谱测量氨基酸含量时,用的OPA柱前衍生化法,可是同样条件下一个样品,测量结果的保留时间,峰面积的重现性很差,峰面积明显不同,保留时间差0.5分钟左右,可是师姐用这台仪器测量藻毒素重现性就没有问题,我可以认为是OPA衍生不稳定导致的吗?会有这种可能吗?那大家一般是如何解决的呢?谢谢了,求救救孩子趴!
本标准规定了鱼类肌肉中主要氨基酸含量测定的试剂与材料、仪器和设备、抽样、分析步骤和结果判定
本人在8月发表的一篇原创中提及”甘氨酸与组氨酸无法分离“的问题,在经过10多天的准备,已有不小的收获,现在分享。摘要 目的: 建立用高效液相色谱法测定人凝血因子VIII中氨基酸含量。方法: 采用6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 为衍生剂,与氨基酸柱前衍生后,用Agilent 1200 高效液相色谱仪,AccQ·Tag C18柱( waters 150 mm ×3. 9 mm,4 μm) ,以水Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液和乙腈进行梯度洗脱,检测波长为248 nm,柱温37 ℃,进样量10μL。结果: 各氨基酸在32 min 内测定完毕,回收率为98.7% ~ 101.5%。RSD 均小于1. 5%。结论: 本法分离度好,快速、简便,可作为产品的质量控制方法。关键词: 6 - 氨基喹啉- N - 羟基琥珀酰亚氨基氨基甲酸酯; 人凝血因子VIII; 甘氨酸; 衍生物; 梯度洗脱; 高效液相色谱法;氨基酸; 含量测定人凝血因子VIII,本品对缺乏人凝血因子礓所致的凝血机能障碍具有纠正作用,主要用于防治甲型血友病和获得性凝血因子Ⅷ缺乏而致的出血症状及这类病人的手术出血治疗。该药物制备过程中使用了氨基酸( 精氨酸、丙氨酸、甘氨酸、组氨酸、盐酸赖氨酸、脯氨酸 等) 做稳定剂,为了保证药品质量和用药安全,应对其中氨基酸的含量进行控制。该法依据过量的6 - 氨基喹啉基- N - 羟基琥珀酰亚氨基氨基甲酸酯( AQC) 在一定条件和氨基酸形成稳定的衍生产物( 柱前衍生) ,用高效液相色谱法测定衍生产物,根据衍生产物的含量计算人凝血因子中各氨基酸的含量。1 仪器和试药1200 高效液相色谱系统( 美国Agilent 公司) ,配置低压四元梯度泵、1314B 紫外吸收检测器、自动进样器、柱温箱、Chemistations 化学工作站; Sartorius CP225D 电子微量天平( 德国Sartorius 公司) ; SartoriusPB - 21 型pH 计( 德国Sartorius 公司) ; LDZ5 -2 低速自动平衡离心机( 上海医用离心机厂) 等。各标准品均来自于中国食品药品检定研究院2 色谱条件及系统适用性试验色谱柱: Waters AccQ·Tag C18色谱柱( 3. 9 mm ×150 mm) ; 流动相: 水为溶剂D,Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液( A) - 乙腈( B) - 水( D) ,柱温:37 ℃; 检测波长: 248 nm。精密量取对照品溶液与供试品溶液10 μL,分别注入液相色谱仪,记录色谱图32 min。3 溶液制备3. 1 Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液称取三水乙酸钠190. 4 g,加注射用水1000 mL,搅拌,溶解,用稀磷酸将pH 调至5. 2,加入乙二胺四乙酸二钠溶液( 称取乙二胺四乙酸二钠100 mg,加注射用水100 mL,摇匀使其溶解) 10 mL,加入叠氮化钠0. 1 g 及三乙胺23. 7 mL( 17. 2 g) ,用稀磷酸滴定至pH 4. 95,用0. 45 μm 的滤膜过滤,于4 ℃储存,备用( 此条件下可保存6 个月) 。量取该溶液100 mL,加注射用水稀释至1000 mL,混匀,即得Eluent( 醋酸盐- 磷酸盐缓冲液) 稀释液。3. 2 对照品储备液混合对照品储备液精密称取各氨基酸对照品适量,置同一100 mL量瓶中,以注射用水溶解并定容至刻度。制成含氨基酸含量均含5. 0 mg·mL - 1 的混合对照品溶液,即得。单个对照品储备液: 精密称取各含氨基酸的各对照品适量,分别置100mL 量瓶中,用注射用水溶解并定容至刻度。制成分别含各氨基酸的单个对照品溶液,即得。3. 3 供试品储备液3. 3. 1 加样回收率试验溶液精密称取各氨基酸各0. 3200,0. 4000,0. 4800 g 和辅料适量,加人凝血因子VIII原液7. 5 mL,肝素钠适量,用1. 0 mol·L - 1 盐酸调pH 至6. 9,加0. 01 mol·L - 1枸橼酸三钠溶液溶解并定容于20 mL。分别制备成16. 0, 20. 0, 24. 0 mg·mL - 1溶液。3. 3. 2 空白溶液 按公司处方,加入辅料的混合物,用注射用水制备各空白溶液3. 4 内标溶液精密称取α - 氨基丁酸( AABA)0. 4 g,加注射用水定容至100 mL。4 氨基酸衍生方法4. 1 精密量取供试品储备液、样品及对照品储备液各1. 0 mL,加1. 5%磺基水杨酸9. 0 mL,混匀静置2 h以上, 3000 r·min - 1离心10 min,留取上清液。4. 2 精密量取“4. 1”项下上清液1. 0 mL( 其中对照品储备液对应上清液分别精密量取0. 06, 0. 4,0. 8,1. 0, 1. 2, 1. 6 mL) ,分别置10 mL 量瓶中,用注射用水定容。制备成供试品溶液、样品溶液及浓度分别为1. 5, 10. 0, 20. 0, 25. 0, 30. 0,40. 0 mg·mL - 1 的对照品溶液。4. 3 精密量取“4. 2”项下溶液各100 μL,分别加注射用水0. 4 mL 及内标溶液20 μL,混匀备用。4. 4 精密量取“4. 3”项下溶液30 μL 放入衍生管中,加硼酸缓冲液( pH 8 ~ 10) 210 μL 涡旋混合,并加入AQC 衍生剂60 μL 涡旋混合15 s,即为各供试品溶液,待用。







 我要推广仪器
我要推广仪器
 下载APP
下载APP