可爱宝贝
第1楼2010/09/20
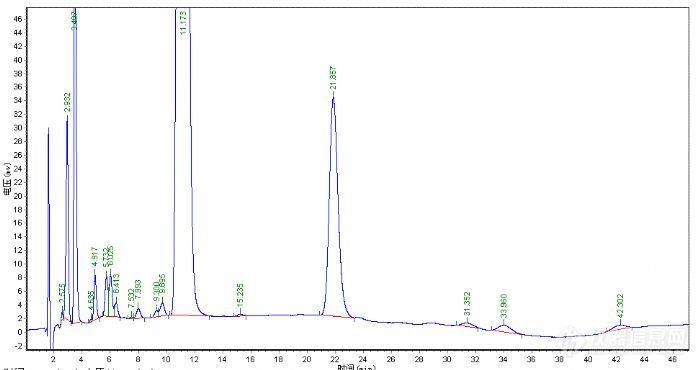
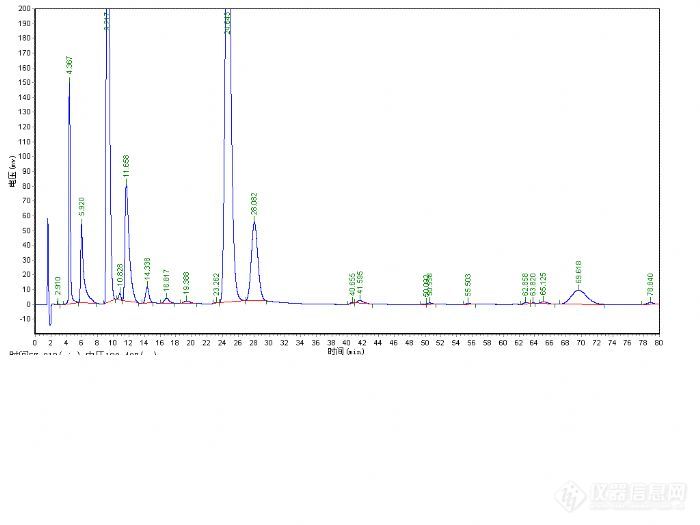
由于没有现成的方法,所以第一步是找到一根合适的色谱柱,从厂家提供的谱图中看出主成分的保留时间在17min左右,初步分析应该是一根250mm的色谱柱,但7-14min这段时间没有组分流出,并且16min前组分分离度都比较好,就决定用一根150mm的色谱柱试试,如果主成分分离不好可以试试梯度洗脱。有了大概的思路后,就从现有的色谱柱里寻找色谱柱来进行调试。刚好两个月前才在月旭购买了一根SB-C18 3um*4.6*150m的色谱柱,就用它了,HPLC选择了岛津的LC-10AT N2000工作站说动手就动手,那就先用甲醇和水去试试吧。检测波长就用紫外进行了全波长扫描,确实在220nm左右吸收波长比较大,因此检测波长确定在220nm。
以下是分析条件
流动相为甲醇:水(体积比50ml:50ml)
流动相稀释样品
进样量:20ul
分析时间:60min
柱温:(℃)25
检测波长:220 nm
流速:1.0ml/min
类型:恒流
检测器:紫外 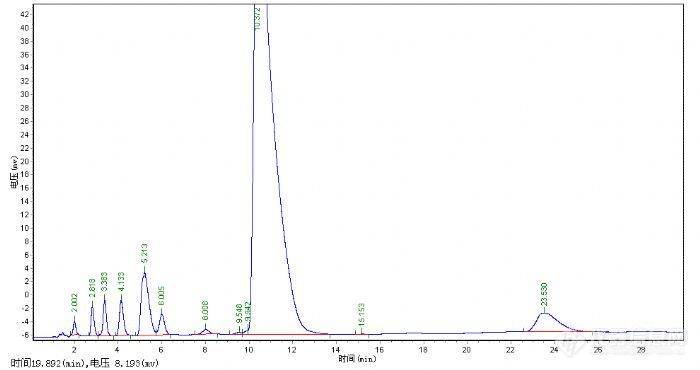
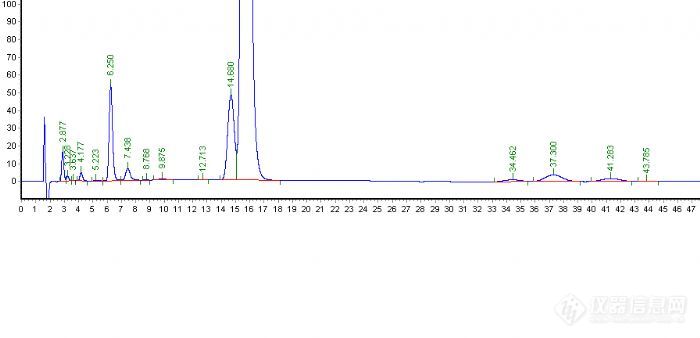
从峰型上看这种方法肯定是不可取了,保留时间是提前了,可拖尾这么严重。下面的工作是如何改变拖尾峰的问题了,曾经看到月旭版面介绍分析方法中提到用酸和碱可以改善峰型,那么就加点三乙胺和冰乙酸看看会不会有所改变。
可爱宝贝
第5楼2010/09/20
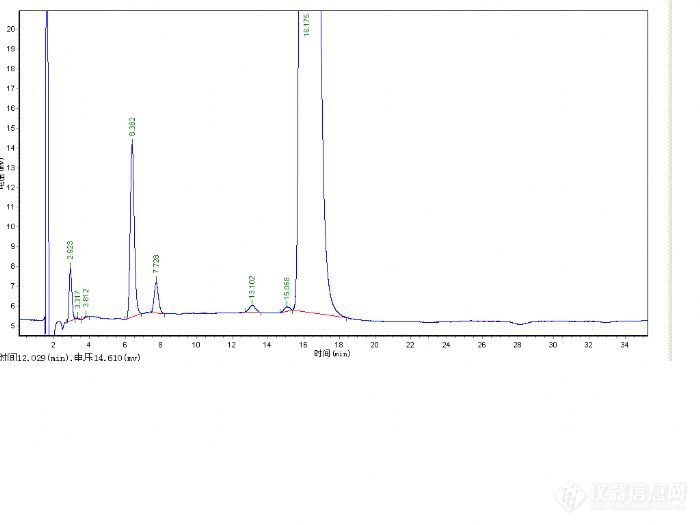
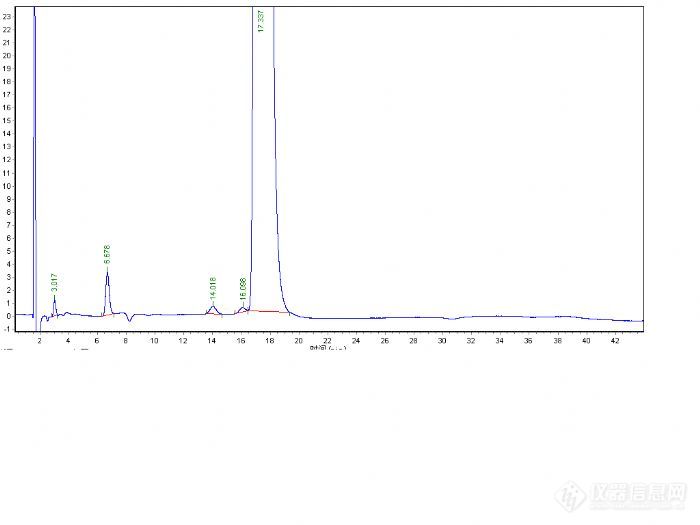
总结一下:这次方法调试总得来说比较顺利,也可能是运气比较好,碰对了。下次碰到类似问题,首先要确定检测波长,波长不正确很难保证数据准确,导致误判。
其次流动相的选择能用甲醇不用乙腈,呵呵从成本考虑,以前也听说先用100%的甲醇先走一遍,看看保留时间,再慢慢调整,这次首先看到客户的谱图组分的保留时间比较散,用100%甲醇肯定不行,所以用了50:50的比例,然后看保留时间再进行调整。
再次,峰型不好可以添加一些酸或碱调整峰型。
最后一点就是要看样品重复性、稳定性,采取相应措施来预防,有的样品怕光,有的怕高温,有的固体稳定,液体不稳定,必须找到使样品分析结果又很好的重复性的方法才行。
以上是本人的个人看法,也许有不对或一知半解的地方,请各位专家指正为好。