

能谱学习报告
9月16日于苏州
10。
 采集角,采集角也叫立体采集角,由取出角和探头到样品的距离共同决定。采集角是直接决定CPS的,对于我们的能谱仪来说探头到样品的距离是固定的,而T一般不变化,所以直接决定CPS的是WD。我们的标准WD是10mm。
采集角,采集角也叫立体采集角,由取出角和探头到样品的距离共同决定。采集角是直接决定CPS的,对于我们的能谱仪来说探头到样品的距离是固定的,而T一般不变化,所以直接决定CPS的是WD。我们的标准WD是10mm。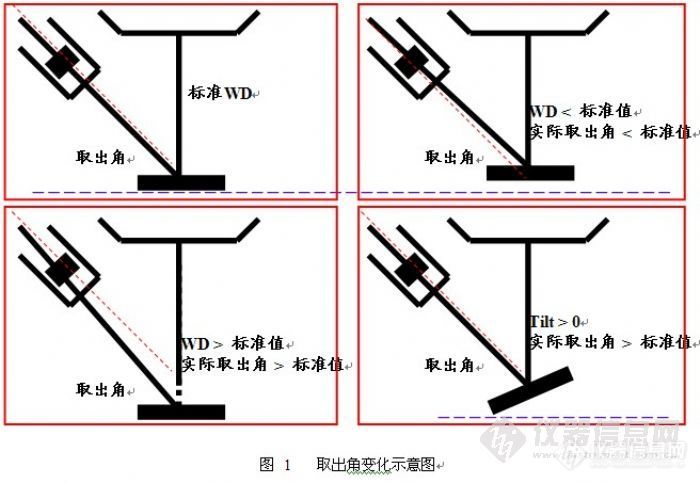


附件:
linzq
第5楼2010/11/01
拿下来仔细看了一下,有些观点很赞成,有些很有启发,有些有点不同看法,写出来大家讨论。没有讨论没有提高。
2.1 过压比
定义:过压比是指SEM所加的高压与某元素的某线系之最低激发能(E0)之比。
例如:Al的Kα的激发能E0=1.48,则当加速电压为15KV时,过压比10。
典型的加速电压应该介于合金中最重元素的能量的2倍和最轻元素的能量的10~20倍。
这点很同意,我在做测试时也有这样的感觉。
低过压比意味着在谱线中产生小的,差的激发峰和较差的统计学品质,而过高的的过压比会使相应元素的能谱结果含量偏高,偏高程度与过压比成正比。这就是我们做能谱定量分析中,H元素直接测出的结果质量百分比严重偏高,因为在20KV下其过压比高达80。
这里有点不同的观点。H元素由于只有一层电子所以没有电子的跃迁等,因此也就没有特征X射线的激发而没有能谱的测量结果。能谱测量范围是BE-U
一般来说选择10倍过压比用于定量分析,20 倍过压比用于定性分析。高压下做定性分析,根据结果再酌情做定量分析。
这点很有启发的我要在今后的测试中好好的体会一下
对于我们的试样,推荐20KV的加速电压,特殊情况另做分析。
这点很同意
2.2 取出角(take off angle)
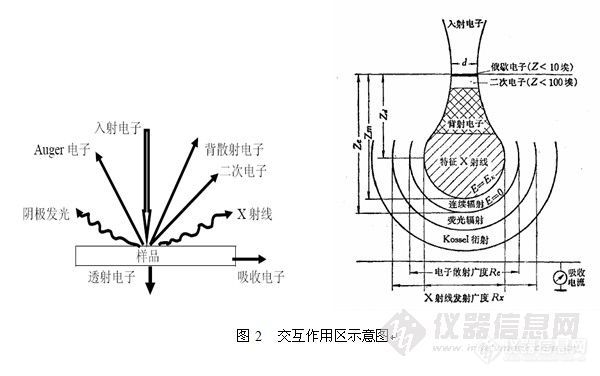
2.3 交互作用区
交互作用区是用来说明X射线信号在样品内部的运动轨迹的统计学特征。如图2所示,为其示意图,右图中梨状的部分就是交互作用区。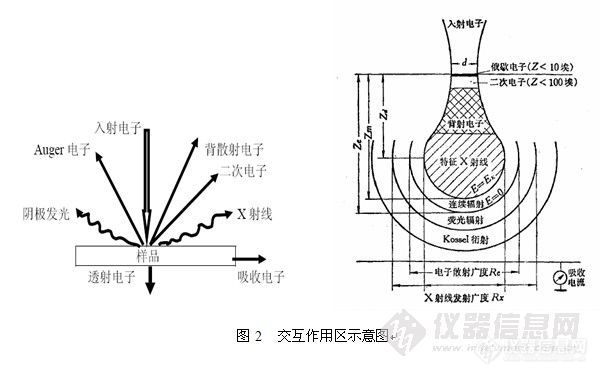
如图所示,二次电子产生区域深度小于10nm,背散射电子产生区域深度约为1~2μm,X-射线和阴极荧光产生区域介于2~5μm。
这里应该提“溢出深度”比较合理一些
入射电子产生的相应物理信号的深度决定其图象的分辨率,所以在各种信号图象的分辨率上,二次电子>背散射电子>特征X射线。另外,做EBSD时产生的X-射线并非图中的X-射线。
EBSD不是用的X-射线,是用背散射电子来做衍射花样
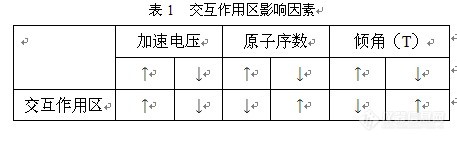
影响因素,影响交互作用区大小的因素有加速电压、样品的平均原子系数和样品倾斜角度(T)。在只有一个变量变化时其变化规律如表1所示。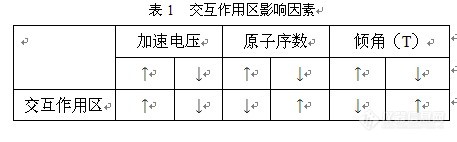
理解此概念对正确分析微小尺寸颗粒与超薄层厚材料非常重要,例如薄膜材料或者做失效分析。经常提到的束斑尺寸与交互作用区无关。

































