不同标椎中喹诺酮类兽药残留检测标准解读
摘要:喹诺酮类药物为两性化合物,本文针对它们的两性结构从喹诺酮类所涉及的比较典型的检测方法标准(如GB/T 21312-2007动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法、GB/T 20366-2006 动物源产品中喹诺酮类残留量的测定 液相色谱-串联质谱法、GB/T 23412-2009 蜂蜜中19种喹诺酮类药物残留量的测定方法 液相色谱-质谱质谱法、农业部1077号公告-1-2008 水产品中17种磺胺类及15种喹诺酮类药物残留量的测定 液相色谱-串联质谱法、SN/T 1751.2-2007 进出口动物源食品中喹诺酮类药物残留量检测方法 第2部分:液相色谱-质谱/质谱法、GB/T 20751-2006 鳗鱼及制品中十五种喹诺酮类药物残留量的测定 液相色谱-串联质谱法)以及最新实施标准:GB 31656.3-2021 食品安全国家标准 动物性食品中四环素类、磺胺类和喹诺酮类药物残留量的测定 液相色谱-串联质谱法这几个标准分别从提取溶剂、净化方法、定量方式、方法定量限讲述喹诺酮类兽药残留检测注意事项。
关键词:喹诺酮类;相关标准;提取溶剂、净化方法
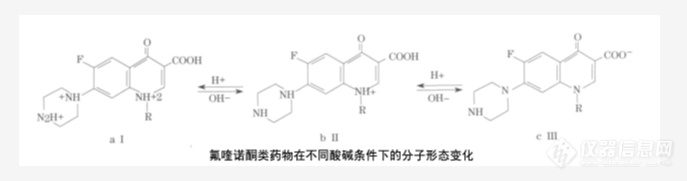
喹诺酮类药物分子结构(以恩诺沙星、环丙沙星为例):氟喹诺酮类药物的结构形态受酸碱度的影响非常显著。在酸性介质(pH=3~5)中氟喹诺酮以两性离子(分子形态)存在于溶液中;当pH为中性时,氟喹诺酮为弱阳离子形式存在;在碱性条件(pH=12~13)时氟喹诺酮为阴离子形式存在于溶液中,正是由于喹诺酮类化合物的两性结构性质决定既可以用酸性溶剂、中性溶剂及碱性溶液来提取。
GB/T 21312-2007动物源性食品中14种喹诺酮药物残留检测方法 液相色谱-质谱/质谱法

提取溶剂:用0.1mol/L EDTA与磷酸氢二钠、柠檬酸组成的pH=4.0的缓冲液提取样品中的喹诺酮类药物。
净化方式:用HLB亲水亲油固相萃取柱(要求目标物以分子形式存在)净化。用甲醇、水活化,提取样液过柱,弃去样液,用5%甲醇水溶液淋洗,弃去淋洗液,抽干,用6%甲醇洗脱并收集洗脱液。
定量方法:空白基质外标法定量,曲线范围(2.5~100ng/mL)
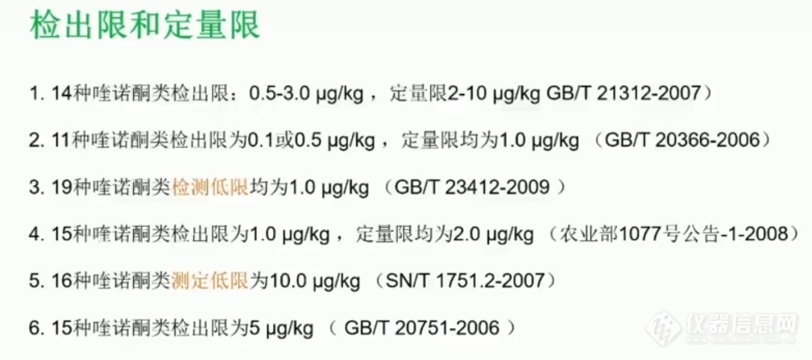
检出限和定量限:14种喹诺酮检出限0.5~3.0μg/kg,定量限2~10μg/kg。
GB/T 20366-2006动物源产品中喹诺酮类残留量的测定 液相色谱-串联质谱法 标准解读


提取溶剂:试样中喹诺酮类残留,采用2%甲酸-乙腈提取。
净化方式:提取液用正己烷液液分配萃取净化除脂净化。
定量方法:溶剂(2%甲酸-乙腈(相当于是纯溶剂)不介意使用,要注意溶剂效应的问题;检测时改用20%乙腈水作为稀释标准溶液的溶剂)外标法定量,曲线范围(5.0~500ng/mL)
检出限和定量限:11种喹诺酮检出限0.1或0.5μg/kg,定量限均为1.0μg/kg。
GB/T 23412-2009 蜂蜜中19种喹诺酮类药物残留量的测定方法 液相色谱-质谱质谱法标准解读

提取溶剂:用0.1mol/L氢氧化钠溶液(pH=13)溶解蜂蜜样品。
净化方式:离子化的喹诺酮类残留物经过阴离子交换固相萃取柱(PAX柱)富集净化;依次用3mL甲醇和3mL水活化固相萃取小柱,将样品溶液转移到小柱上,依次用水、甲醇淋洗,弃去上述滤液。用5%甲酸-甲醇溶液3mL洗脱,收集洗脱液。
定量方法:溶剂(20%甲醇水)内标法定量,曲线范围(1~60ng/mL)
检测低限:1.0ug/kg。
农业部1077号公告-1-2008水产品中17种磺胺类及15种喹诺酮类药物残留量的测定 液相色谱-串联质谱法 标准解读

提取溶剂:用1%酸化乙腈提取样品中的喹诺酮类化合物。
净化方式:正己烷液液分配萃取除脂肪净化。
定量方法:溶剂(20%甲醇水),内标法定量,曲线范围(10~200ng/mL)
检出限和定量限: 15种喹诺酮检出限1.0μg/kg,定量限均为2.0μg/kg。
SN/T 1751.2-2007 进出口动物源食品中喹诺酮类药物残留量检测方法 第2部分:液相色谱-质谱/质谱法 标准解读

提取溶剂:用1%酸化乙腈提取样品中的喹诺酮类化合物。
净化方式:正己烷液液分配萃取除脂肪净化。
定量方法:空白样品基质外标法定量,曲线范围根据样液浓度待定。
测定低限:10.0ug/kg。
GB/T 20751-2006 鳗鱼及制品中十五种喹诺酮类药物残留量的测定 液相色谱-串联质谱法 标准解读

提取溶剂:试样中残留的喹诺酮类药物采用乙腈提取。
净化方式:提取液经正己烷液液分配脱脂后,以强阳离子固相萃取小柱(SCX)净化:用前依次以3mL甲醇、3mL水、3mL10mmol/L乙酸铵缓冲液(pH4.6)活化,保持柱体湿润。样液经正己烷液液分配脱脂,氮吹干,以3mL10mmol/L乙酸铵缓冲液(pH4.6)溶液上SCX柱,以约1mL/min的流速全部通过强阳离子交换柱,抽干,先后以1.5mL甲醇、3mL 25%氨水-甲醇洗脱,合并洗脱液。
定量方法:空白样品基质外标法定量,曲线范围根据样液浓度待定。
测定低限:15种喹诺酮类均为5.0ug/kg。
GB 31656.3-2021食品安全国家标准 水产品中诺氟沙星、环丙沙星、恩诺沙星、氧氟沙星、噁喹酸、氟甲喹残留量的测定 高效液相色谱法标准解读
适用范围:水产品中鱼类肌肉组织,虾、蟹、贝类的可食组织中诺氟沙星、环丙沙星、恩诺沙星、氧氟沙星、噁喹酸、氟甲喹残留量的检测。
提取液:用1% 酸化乙腈提取。
净化:C18柱净化,依次用甲醇、水、磷酸盐缓冲液各3mL活化。取样液过柱,流速控制为每秒1滴。水3mL淋洗,抽干,加洗脱液5mL(25% 氨水甲醇液),收集洗脱液。
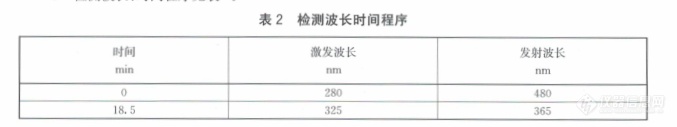
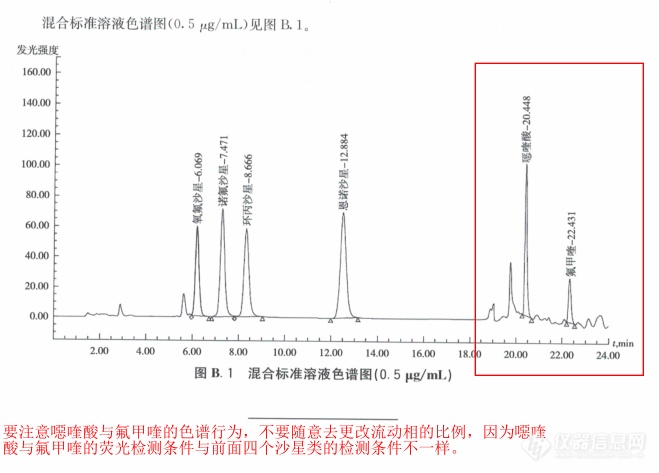
总结:



通过对这几个相关的喹诺酮类标准的检测标准解读,对喹诺酮类药物的提取,可以使用EDTA-Mcllvaine缓冲液(pH4.0),甲酸乙腈(1%~2%)(pH3~5), 纯乙腈(pH= 7),还可以使用0.1mol/L氢氧化钠溶液(pH=13),是因为喹诺酮类药物为两性化合物,不同的pH溶液提取决定了其在溶液中的存在形式不同,如果用pH3~5酸性溶液来提取的话,此时喹诺酮类药物以分子形式存在于溶液中,净化的方式则可以采用过HLB、C18、或正己烷液液分配萃取净化;如果是采用乙腈来提取的话,则喹诺酮类药物以阳离子形式存在于溶液中,因此净化方式要采用阳离子交换柱来净化;如果采用碱性溶液来提取的话,此时喹诺酮类药物以阴离子的形式存在于溶液中,此时则要相应的采用阴离子交换柱来净化。



































