-
+关注
私聊
-

皮皮鱼
第124楼2009/04/12
这个柱子是一个非极性沸点出峰的柱子。如果你的烃类组分主要是烷烃,应该可以。如果烃类中有较多的烯烃炔烃,那么估计不成。
建议用一根wax柱子和一个这种柱子连用,增加醇类保留时间,和烃类得到良好分离,此时炔烃、二烯烃分离效果不好。
当然能够用Deans中心切割就更好,可以用wax和Al2O3柱共用,炔烃都能得到良好分离。
网上有安捷伦的色谱柱谱图大全之类的吧,下载一个,查看各种组分的出峰情况,这个是最关键的。pink007111 发表:yuen72先生您好!我有以下问题想请教,
请问:我想分析CO+H2合成低碳醇的液相产物,主要是烃类和醇(C1-C5)的混合液体,请问RTX-1 的柱子可以分析吗?如果不行需要怎么选择,选择的依据是什么?谢谢
-
+关注
私聊
-
皮皮鱼
第126楼2009/04/12
1、这样做必然带来衍生化效率造成的误差,如果衍生化效率很高,超过99%,可以直接用。如果衍生化很难做到这么高,那么可能必须用酸标准做衍生化处理标定。衍生化过程中产生的线性偏离,应该是衍生化过程中控制不好,导致发生样品损失造成,应该通过控制标准操作步骤来消除。
2、可以。如果衍生化过程和进样都能精确控制体积,那么完全可以用外标替代内标来分析。之所以用内标,可能是方法本身进样无法控制准确,液体样品一般用注射器进样,体积精确性差一点。现在用自动进样器,体积控制能力高的多了。wangsht4 发表:yuen72先生您好!我有以下问题想请教,
请问:最近我们在做生活饮用水中的二氯乙酸和三氯乙酸,按照国家标准GB5750-2006生活饮用水标准检验方法,需要采用甲醇酯化后进行测定,然后添加1,2-二溴丙烷作为内标,采用内标法定量
我们现在采用进口的衍生化好的标准样品(二氯乙酸甲酯和三氯乙酸甲酯)基本确定了两种物质的保留时间,下一步准备做标准工作曲线
有两个问题想请教一下:
1 做标准工作曲线的时候能够采用二氯乙酸甲酯和三氯乙酸甲酯做而不进行衍生化预处理?
如果用二氯乙酸甲酯和三氯乙酸甲酯直接做标准工作曲线,这样得到的曲线线性可能会好些,但可能会引入系统偏差;而如果用二氯乙酸和三氯乙酸衍生化然后在做标准工作曲线,这样得到的曲线线性不一定好,但可以基本忽略掉衍生化带来的系统偏差
2 能否用外标法代替内标法做标准工作曲线?
-
+关注
私聊
-
皮皮鱼
第128楼2009/04/12
质谱接触很少。我觉得内标这个方法可以采用,但不能用同一个校正因子。相对校正因子都是可查的,利用出峰时间相近这个内标的办法来标定就可以了。例如乙酸乙酯和丁酸乙酯可能距离比较远,而丁酸与它距离近,可以用丁酸来内标即可。MS的MC图,和你选取的离子有关,如果选择好离子,用同类物质内标应该也是可以的。
半定量分析,这么做应该是完全可以的。选取合适的离子碎片,是提高灵敏度、减小误差的有效办法。其他应该都是色谱的问题,进样等等,就不多说了。fantasyk 发表:yuen72先生您好!我有以下问题想请教,
请问:我是用固相微萃取-气质联用定量香气成分,采用内标法.
因为香气成分中种类较多,且大部分物质标准品难以获得,我打算采用多种内标对应同类物质进行校正,如己酸乙酯校正酯类组分,辛酸校正酸类组分,不知道这种方法是否可行?
同时,由于保留时间分布较宽,按您前面讲解的多点内标原理,是否会给内标定量带来不准确性?如果作为半定量,这种不准确性是否在可考范围之内?
除了多点内标(成本因素)外,还有什么办法可以尽量减小上述方法的分析误差.谢谢
-
+关注
私聊
-

迷途小书童
第131楼2009/04/12
也许是我愚钝,不添加内标物怎么算内标法("没有内标物的是内标法")?要加内标物时先确定样品中是否含有与内标物一样的组分,这个好象是GC工作者都知道的,只是您的写法让我费解了,另外,样品中如果含有内标物的话,您认为是内加法,那现在有一样品,组分有3,分别是ABC,我想定A的含量,我可以加A(我想知道B的含量,我可以加B),根据两针的面积折算真实面积,进而计算A的含量,我怎么看怎么是内外综合,您也认为内标法的源头是外标法啊!外标法和内标法都只要进一针,这个内加法进两针只是为了求得响应因子和修正两次进样量,确实与内外两种方法都不同,内加法只是外标与内标的综合,我没有不同意先生的意见,可能各人的想法不同!至少这三种方法我都用过,只是我不知道最后的一个方法还有名称叫"内加法"!
yuen72 发表:内标法的源头是外标法,内加法的源头是内标法。内标法是基于单次进样,进样体积只有一个,因此可以用外表公式推导得到结论的。内加法是基于未添加组分如果面积不变,则总的待测样品进样量相同,利用内标外标两种方法结合得到分析结果的。所有定量方法的起点都是外标法,这是由检测器的线性关系决定的。在前面的讲授中有说明,希望能够仔细看一下。
没有内标物的叫内标法,这个不是笔误,样品中没有内标物,这是内标法的基本要求。当样品中含有内标物的时候,就变成了内加法。
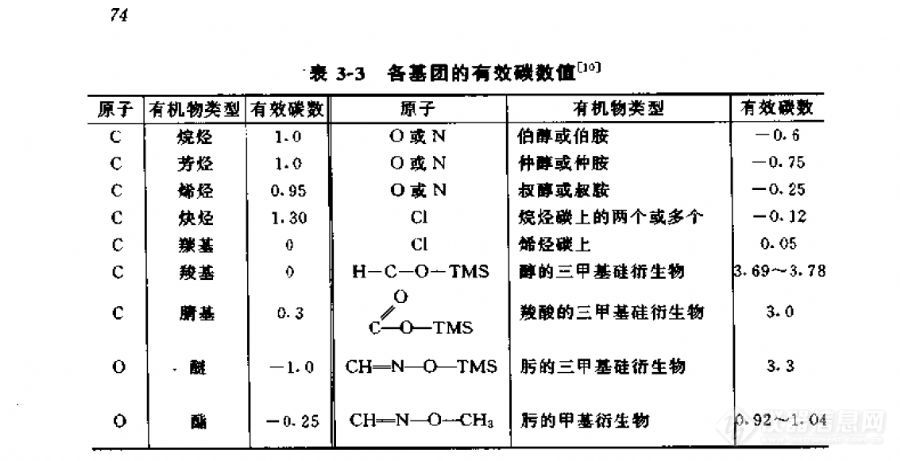
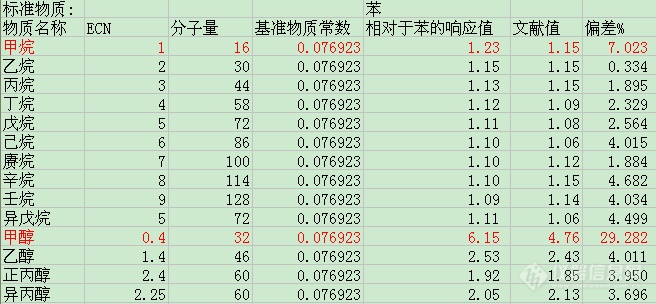
-
+关注
私聊
-
皮皮鱼
第133楼2009/04/13
呵呵,理解并能应用就好。至于如何定义,这是学者的事情,我是只管应用的。内加法,在《化验员读本》(化学工业出版社,刘珍主编,第四版)上这么定义,我接受了。其他书上定义这个方法为“标准加入法”,如《色谱定性与定量》,我也认可的。无论叫什么方法,哪怕叫两次分析法也可以,能够合理应用就成了。
内标法的基本要求如下:1、样品中不含有内标物。2、内标物易得纯物质。3、内标物出峰时间与待测组分接近。4、内标物含量与待测组分含量接近。当然,还有内标物性质要稳定等其他要求,但第一条说的很清楚。我所说的没有内标物,指的是待分析样品中不含有内标物。希望能理解。
样品含有ABC三个组分,内标法的做法是添加D组分,这个D组分必须在你的样品中是没有的。此时仅仅需要添加后样品进色谱分析即可。
据你所言,无论添加A还是B,用我这个讲义的定义(以《化验员读本》为依据),都是内加法,或者说是“标准加入法”了。标准加入法,加A还是B,只要加入一个组分,就能够计算所有ABC三个组分的含量,用的是相对校正因子。此时,必须有原始样品,以及添加后的样品,分两次进入色谱仪分析。
外标法和归一化法的特点是样品可以直接进样,不要前处理添加物质。当然特殊必须进行浓缩等前处理的是另一回事。
内标法的特点是添加一个样品中没有的内标物,才能进行分析。是样品中没有内标物,而不是没有内标物。
内加法的特点是找不到这样的内标物,因此添加了一个样品中含有的物质,因此不仅仅要做添加处理后的样品分析,还要对未处理的样品进行分析。内加法计算中,核心调整不是调整到外标法需要的总进样量相等,而是进样中待分析样品量相等,也就是说,调整了的添加后样品总进样量=调整了的未添加样品进样量 + 添加的物质量。即使经过调整,仍然不是外标法规定的进样稳定。
内加法兼具内标外标的特点,内标具有外标的核心,这个说法我都认可。还是这句话,能够理解,并在分析中合理应用即可。
我在讲座中放了内加法的实例,就是为了增加理解。希望能够明白计算过程中每一步的含义。lvqiang_2001 发表:也许是我愚钝,不添加内标物怎么算内标法("没有内标物的是内标法")?要加内标物时先确定样品中是否含有与内标物一样的组分,这个好象是GC工作者都知道的,只是您的写法让我费解了,另外,样品中如果含有内标物的话,您认为是内加法,那现在有一样品,组分有3,分别是ABC,我想定A的含量,我可以加A(我想知道B的含量,我可以加B),根据两针的面积折算真实面积,进而计算A的含量,我怎么看怎么是内外综合,您也认为内标法的源头是外标法啊!外标法和内标法都只要进一针,这个内加法进两针只是为了求得响应因子和修正两次进样量,确实与内外两种方法都不同,内加法只是外标与内标的综合,我没有不同意先生的意见,可能各人的想法不同!至少这三种方法我都用过,只是我不知道最后的一个方法还有名称叫"内加法"!