-
+关注
私聊
-

yhl-87_
第21楼2009/11/05
农药残留检验方法系列讲座(21)
呋喃丹-敌百虫混合颗粒剂的气相色谱分析
一、 前言
呋喃丹-敌百虫混合颗粒剂 (简称:呋敌混剂),是以呋喃丹和敌百虫为有效成分的颗粒剂。敌百虫的化学名称为0,0-二甲基-(2,2,2-三氯-1-_羟基乙基)膦酸酯,呋喃丹的化学名称为2,3-二氢-2,2-二甲基-7-苯并呋喃基-N-甲氨基甲酸酯。从呋喃丹与敌百虫的化学结构看来.敌百虫的化学极性比呋喃丹的化学极性大得多,而呋喃丹的熔点比敌百虫的熔点高得多。并且它们都有热分解的性质,呋喃丹在色谱柱中受热后分解为7-羟基呋喃,敌百虫受热后部分分解为敌敌畏,因此给气相色谱定量分析带来一定困难。本研究为的是在同一特定色谱参数操作条件下,使两个化合物在色谱分析过程中既不产生热解 (或者即使有微量热解,亦成为一个恒定常数),而又有较好的分离度、线性关系和满意的定量分析结果。为此对固定相、流动相、温度和检测器类型等进行了实验,并选择了最佳色谱操作条件,使呋哺丹与敌百虫都能获得准确、快速定量分析。该方法的相对误差呋喃丹为2.2%、敌百虫为1.5%。
本文由沈阳化工研究院农药四室的李达潮和耿建文早期撰写的论文,但很有参考价值,现全文介绍如下。
二、 实验部分
(一) 主要试剂及仪器
呋喃丹的纯样:熔点l53~154℃,经与FMC标准样核对,无色谱杂峰,(本院物化室提供);敌百虫纯样:熔点82.5~83.5℃,无色谱杂蜂,(本室提供);内标物正十六烷为化学纯 (天津化学试剂厂); 固定液为聚甲基醚氧烷;固定相为Chromosorb W AW DMCS(60~80目);苯为分析纯;丙酮为分析纯。
SP-501型气相色谱仪-热导检测器 (鲁南化工机械厂产);电动振荡器 (上海市南汇县横沔农机厂产);离心机;微量注射器 5~10微升。
(二) 测量方法
1.色谱仪操作条件
色谱柱:聚甲基硅氧烷固定液6~20%,酸洗担体94~80%,1米长、直径4毫米不锈钢柱,柱温:160℃(表值),汽化温度:160℃ (表值),检测室温度:210℃ (表值),载气:H2.,流速38格 (表值) ,纸速:1200毫米/小时,进样量:5~l0微升
2.定性试验
采用标准呋喃丹、敌百虫纯品和正十六烷内标进行混合,以苯:丙酮(4:1)溶解,在稳定的色谱操作条件下,进样分析,测得各组分的保留时间如图-l。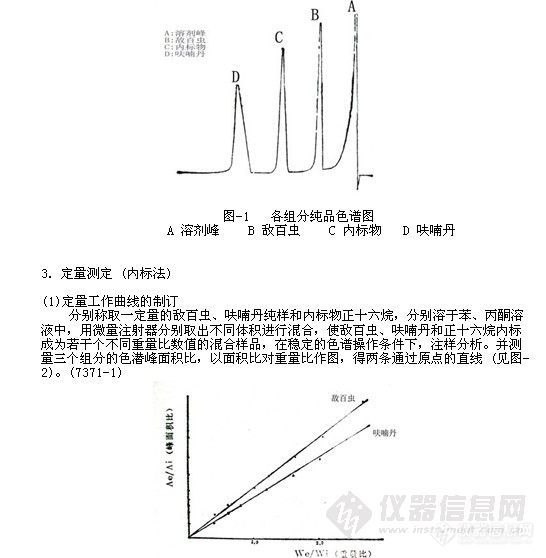
图-2定量工作曲线图
从定量工作曲线看来,呋喃丹、敌百虫对内标物的重量比从0.4到2.5的范围内均有良好的线性关系,采用检量线法定量是可靠的。
4.样品的测定
准确称取一定量的被测样品和内标物,经用溶剂和振荡机萃取后,在稳定的色谱操作条件下进行分析。由色谱图上分别测量出呋喃丹、敌百虫与内标物的峰面积比 (A样/A内),然后从检量线中查出对应的重量比 (W样/W内)。被测物的有效成分百分含量按下式算得。
W×W内
呋喃丹或敌百虫% = ———— ×100
W样
式中:W——在检量线中查得的重量比
W内——内标物正十六烷的重量
W样——被测样品的重量
三、 实验结果
(1) 实测结果
我们首先对院物化室提供的呋喃丹标样、敌百虫标样进行实验测定,结果见图-3和
图-4。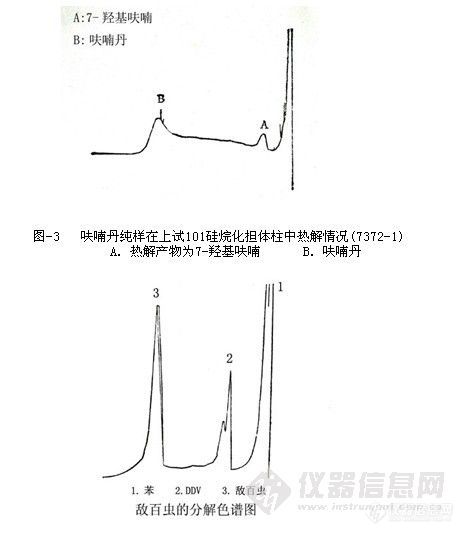
图-4 敌百虫纯样在氮气为载气时分解情况 (7373-1)
1.敌百虫 2.热解产物滴滴畏 3.苯
⑵ 表-1 呋喃丹、敌百虫混剂样品分析结果及偏差表 (7374-1)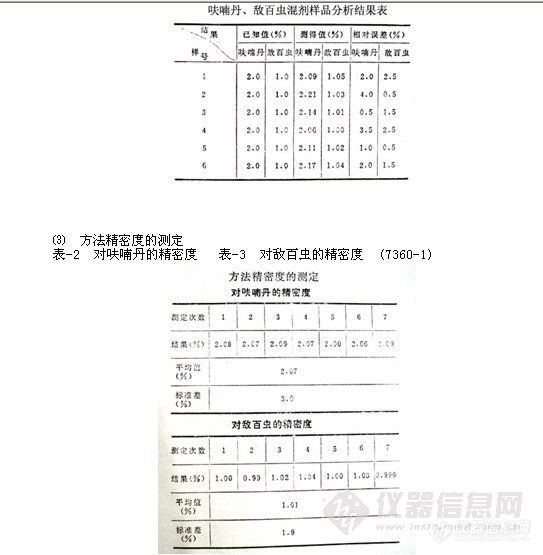
⑷ 回收率的测定
采用已知分析结果的样品,加入一定量的纯样和内标物,以同样方法进行溶剂振荡萃取,在稳定的色谱操作条件下,进行分析,测得结果见表-4。(7376-1)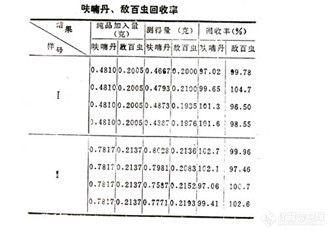
四、 讨论与结论
呋喃丹和敌百虫两个化合物都具有热分解的特性,特别在高温和碱性介质中,即使是弱碱性,也会产生热解,所以在色谱分析过程中,要注意被测物介质的pH值是十分必要,酸性介质比较稳定。另外我们发现在分析过程中,导致呋喃丹与敌百虫热解产物色谱峰的出现与担体、固定液和流动相类型有关。凡属催化性强、吸附性大的担体,_一些酯类固定液以及线速度低的载气,都会促使被测物在色谱柱中产生热解。实验表明上试l01硅烷化白色担体对被测物就有明显的热解 (见图-3)。采用氮气作载气时,由于氮气的线速度小.热导系数低,在分析过程中。敌百虫有明显的热解(见图4)。
然而本文的色谱操作参数进行呋、敌混剂的分析,基本上避免了呋喃丹与敌百虫在色谱柱中的热解现象出现。其结果可以从定性色谱图 (图-1)中看出既无7-羟基呋喃峰,也无敌敌畏峰。
本研究的定量方法是采用内标检量线法,为此在分析时,必须严格控制与制订检量线时相同的色谱操作条件。但是本文的色谱操作条件亦适用于内标和外标法(插入法),即采用校正因子(f值)进行定量,我们曾对两种定量方法进行比较,都获得一致结果。
本方法测定呋、敌混剂的有效成分含量,无沦从分离度,还是从线性关系来看均较理想,敌百虫的标准差为3.0%,呋喃丹的标准差小于2%。各组份的出峰时间仅4分钟,方法达到快速、准确的要求,适于工业生产分析。
-
+关注
私聊
-
yhl-87_
第22楼2009/11/05
农药残留检验方法系列讲座(22)
农药残留检验方法系列讲座
氨基甲酸酯类农药的毛细管气相色谱法
㈢ 毛细管气相色谱法
1、毛细管柱对呋喃丹及其代谢物的灵敏度
毛细管柱可以排除杂峰干扰,分离多种氨基甲酸酯农药,分离度明显提高(见图-1),同时便于添加筛选内标物。
图-1为SE-54毛细柱程序升温图(条件:30m×0.25mm×0.25μm,)柱温80℃(1min)以10℃/min升至230℃(20min).。
图-2为2.5%QF-1+ 5%DC-200(1.75mm×2m Gas Chrom Q)填充柱程序升温140℃(2min)以 (1min)再以10℃/min升至210℃(5min)。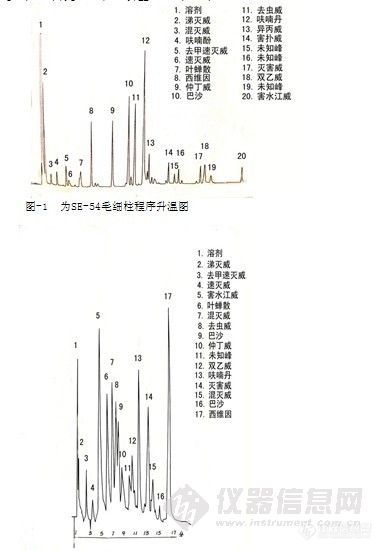
图-2 填充柱程序升温
表-1 OV-17毛细管柱对呋喃丹及代谢物的灵敏度表
* 计算值
2、毛细管柱对常见农药的分离
能分析分离农药的毛细管柱有(一般选用非极性、弱极性和中极性柱):
如
1、SE-30 非极性(也称为OV-1、DB-1、BP-1、RTX-1、007-1、HP-1等)
2、SE-54 非极性(也称为OV-73、DB-5、BP-5、RTX-5、007-2、HP-5等)
3、DB-1301 弱极性(也称为DB-624、RTX-1301、RTX-624、HP-1301等)
4、DB-35 中极性 (也称为RTX-35、SPB-35、HP-35等)
5、DB-1701中极性 (也称为RTX-1701、HP-1701等)
6、OV-17中极性 (也称为DB-17、RTX-50、HP-50+、HP-17、007-17、SP-2250
等)
以上六种不同极性的毛细柱均能分析分离多种农药残留,现举一例供参考: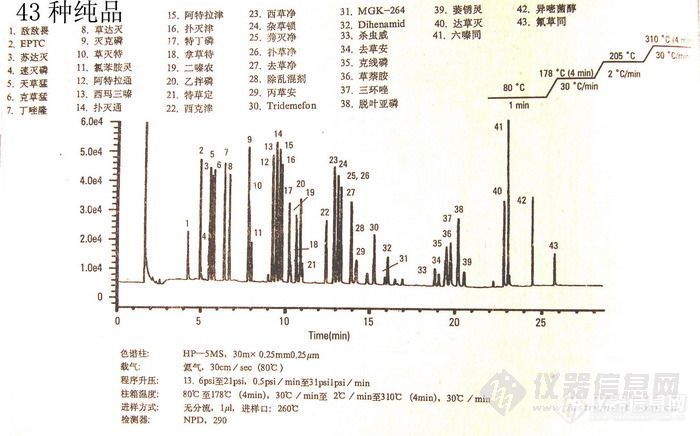
图-3 43种农药纯品毛细管气相色谱分离图
使用上面介绍的2号柱 HP-5MS(指质谱专用柱),柱温80℃(1min),以30℃/min升至178℃(4min),再以2℃/min升至205℃,最后以30℃/min升至310℃(4min)。分离效果不错,对43种农药纯品放大观察均能分离开。
-
+关注
私聊
-
yhl-87_
第23楼2009/11/05
农药残留检验方法系列讲座(23)
气相色谱-质谱法同时测定水中
7种氨基甲酸酯类杀虫剂
1、前言
氨基甲酸酯类杀虫剂是继有机氯、有机磷之后出现的一类重要杀虫剂.具有作用迅速、选择性高、易分解、残留毒性小等优点,而被广泛使用。但随着此类杀虫剂使用范围的不断扩大.大气、水体、土壤等环境遭到不同程度的污染,由其引起的食物中毒、误服以及投毒事件时有发生.因此有必要建立一种多种氨基甲酸酯类杀虫剂的分析检测方法。为环境检测和临床诊断提供依据。氨基甲酸酯类杀虫剂的测定方法主要有薄层层析法、气相色谱法、液相色谱法⑴ ⑵、液相色谱-质谱法、气相色谱-质谱法⑶-⑻等,但未见GC—MS同时测定该7种氨基甲酸酯类杀虫剂的报道。
本文是由成都疾病预防控制中心的高玲、杨元、王炼及成都市成华区疾病预防控制中心的李仲祥等研究人员共同完成的,他们用气相色谱一质谱法同时测定水中7种氨基甲酸酯类杀虫剂,曾用于井水、自来水、矿泉水、纯净水等水样中微量氨基甲酸酯类杀虫剂的检定,结果满意。下面全文介绍如下:
2、材料与方法
1-1试剂与仪器
氨基甲酸酯类杀虫剂标准溶液:准确称取呋哺丹、西维因、速灭威、残杀威、仲丁威、异丙威、抗蚜威等7种杀虫剂标准品(购自Sigma公司).用甲醇配成标准储备液。置冷暗处密闭保存.临使用时稀释成单一和混合标准使用液;
甲醇:色谱纯;
ENVI—l8小柱:300mg/3mL(美国SUPELCO公司);
固相萃取装置,带采样器(美国SUPEI;CO公司)
GC-MS: HP6890 GC/5973 MSD;
色谱柱:.HP-35MS毛细管柱(30m×0.25mm×0.15μrn)
1-2 实验方法
⑴ GC—MS分析条件
色谱:柱温为80℃(1min)以10℃/min升至240℃再以100℃/min升至270℃;进样口:280℃,脉冲不分流。脉冲压力为45psi。进样量:lμL。载气流速:1.5ml/min,线速度:45cm/sec。
质谱:EI源,电离能量为70eV。传输线为280℃。全扫描(Scan)测定方式的扫描范围为
m/z 50~5500选择性离子萃取(SIM)测定方式(扫描离子):速灭威:108;异丙威12l、l50;仲丁威;12l、l36;残杀威ll0;呋喃丹:164;抗蚜威166;西维因144。
⑵ 样品处理
取1000ml水样于烧杯中,将水样和固相萃取装置通过采样器连接,真空泵抽滤,使水样通过ENVI—l8小柱,让待测物吸附在小柱上;然后用甲醇洗脱小柱,定容至1ml,GC/MS测定。
⑶ 测定 按⑴设定分析条件,仪器工作稳定后,直接进样测定。
3、 结果与讨论
2-1 固相萃取条件优化
⑴ 萃取流速和洗脱流速的选择 实验表明,加标水样通过小柱的流速在7.5ml/min以下时,实验可以获得比较满意的回收率;洗脱液通过小柱的流速在0.6ml/min以下时,实验可以获得比较满意的回收率。本文选择水样流速为7.5ml/min。洗脱液流速为0.6rnl/min。
⑵ 洗脱液的体积选择 实验发现,以甲醇洗脱小柱时,分别使用0.8 、0.4、0.4mL甲醇分3段洗脱(每1次均抽至无液体流出时,再补加甲醇),最后定容为1mL实验可获得比较满意的回收率。
2-2 GC-MS条件优化
⑴ 色谱柱的选择
实验发现,中等极性的HP-35MS毛细管柱对7种氨基甲酸酯类杀虫剂能实现基线分离,见图-1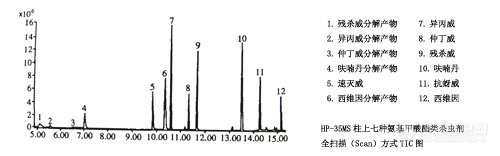
图-1 HP-35MS柱上7种氨基甲酸酯类杀虫剂全扫描方式TIC图 7468-1
⑵ 柱温箱温度优化
氨基甲酸酯类杀虫剂的热稳定性较差,如果色谱柱起始温度太高易发生分解⑤,但起始温度太低,分析时间又过长。经实验考察,本文优化柱温箱温度为80℃ 保持1min以10℃/min升温至240℃,再以100℃/min升温至270℃。
⑶ 进口温度优化
实验表明,当进样口温度为280℃时,所得色谱峰灵敏度最高,色谱峰分离良好。本文选择进样口温度为280℃。
⑷ 进样方式及进样口压力优化
实验表明,当进样口脉冲压力为45psi时所得色谱峰尖锐、灵敏度最高。本文选择进样口脉冲压力为45psi。
⑸ 柱流速优化
实验表明,当载气流速为1.5mL/min时,混合标准的分离效果最好,所得色谱峰灵敏度较高。本文选择载气流速为1.5mL/min。
2-3 质谱检测方式的选择及方法检出限
实验结果证实,在定量分析时SIM检测方式无论在灵敏度、精密度等方面均优于Scan所包含的大量定性信息又是SIM无法与之相比的。因此,本文在定性时采用Scan检测方式,而定量时采用SIM检测方式。结果见表-1
表-1 方法的标准曲线和检出限 7469-1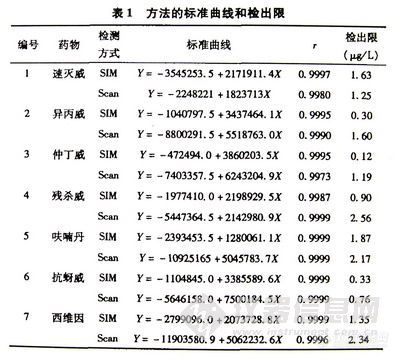
2-4 样品分析
选择不同的水样作为本底,加入7种氨基甲酸酯类杀虫剂混合标准溶液,充分混匀,按前述分析方法进行提取,分别采用全扫描(Scan)测定方式和选择性离子萃取(SIM)方式进行测定,2份加标水平进行5次重复测定,从实验结果看出7种氨基甲酸酯类杀虫剂的回收率分别为:异丙威83.4%-107.7%、仲丁威83.9%-108.5%、残杀威 85.4%-118.0%、呋喃丹82.4%-108.9%、抗蚜威 81.6%-93.9% 、西维因83.6%-94.3%;RSD%在0.55%-10.1%、速灭威为 46.4%-81.9%,符合农药多威留检测的回收率和RSD要求。
参考文献
[1] Bakowski R.S.Gas chromatographic-mass spectrometric confirmation of ten N-methyl earbamati insecticides in liver [J].J AOAC lnt,I994,77(6):1568—1574.
[2] 张洪兰,罗毅.气相色谱法分离检定血中八种氨基甲酸酯 [J].色谱,1994.12(2):117~118,
[3] Yang SS.Recent advances in the residue analysis of N-methyl carbamate pesticides [J].J Chromatogr A,1996,754(1~2):3~16.
[4] Girand D.Determination of traces pesticides in water by solid-phase extraction and liquid chromatography-ionrary mass spectrometri [J].J Chromatogr A,1997,777(1):115—25.
[5] 贾薇,王磊石,赵雷,等.气相色谱-质谱法同时测定七种氨基甲酸酯类杀虫剂 [J].中国卫生检验杂志,2003,13(2):205—206.
[6] Bakowski RS.Gas chromatographic-mass apectrometric confirmation of ten N-methyl carbamate insecticides in liver [J].J AOAC lnt.1994, 77(6 ): 1568~1574
[7] girang D.Determination of traces pesticides in water by solid-phase extraction and liquid chromatography-ionsprary mass spectrometry [J]. J Chromatogr A ,1997,777(1) : 115~154
[8] Climent M J, Miranda MA.Gas chromatographic-mass spectrometric study of photodegradation of carbamate pesticides [J] .J Chromatogr A 1996, 738 (2) : 225~231
-
+关注
私聊
-
yhl-87_
第24楼2009/11/05
农药残留检验方法系列讲座(24)
双毛细管气相色谱法测定水果蔬菜中
有机磷及氨基甲酸酯类农药残留
1 前言
本文采用简便有效的提取方法,以双毛细管柱气相色谱法对果蔬样品中残留的有机磷及氨基甲酸酯类农药进行测定,排除了干扰影响,增加了试验结果的可靠性,取得了较满意结果。
毛细管气相色谱法测定果蔬中农药残留,具有分离效果好,分析时间短,检测灵敏度高等优点⑴。GB/T17331—1998方法样品前处理较为繁琐,且使用单一毛细管柱测定存在一定干扰。现介绍浙江萧山区卫生防疫站的赵立峰和徐陆妹两位完成的分析方法,供大家参考。
2 试验部分
1-1仪器与试剂
Agilent 6890气相色谱仪,附氮磷检测器 (NPD)和HP6890化学工作站,旋转蒸发仪。
丙酮、二氯甲烷为AR级,重蒸。农药标准品浓度均为100mg/L,使用前以丙酮稀释成合适的混合标准使用液。
1-2样品处理
取切碎混匀后的果蔬试样10.0g,置于250mI具塞锥型瓶中,加入无水硫酸钠20~40g(根据果蔬含水量而定),剧烈振荡,再视果蔬色素含量加入活性炭0.2~0.6g脱色。加入丙酮:二氯甲烷(1:1)30mL,振荡提取30min,过滤,再重复提取一次,合并提取后并过滤的提取液。用旋转蒸发仪35℃水浴蒸发至少量,用氮气吹干。以丙酮定容至1.0~5.0mI_。即为样品试液,取2.0μL进色谱分析。
1-3色谱条件
方法一:HP-5,19191J-413石英毛细管柱(3Om×0.32mm×0.25μm);载气为高纯氮气,柱压为114kpa,柱温为90℃,保持0.5min,以15℃/min上升至185℃,再以5℃/min上升至230℃;进样口260℃,不分流进样,吹扫时间0.5min,吹扫流量为100mL/min。;检测器温度290℃,氢气为3mL/min,空气为60mL/min,尾吹气为10mL/min。在上述试验条件下,10种农药得到分离,其中1号与2号(甲胺磷和敌敌畏)7号和8号(乐果和呋喃丹)分离效果差点,见图-1。
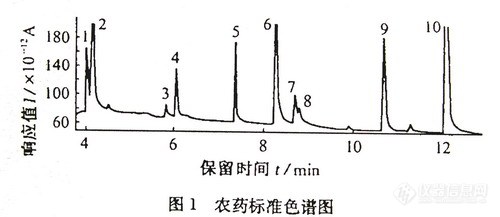
图-l 农药标准色谱图 7551-1
1.甲胺磷(Methamidophos)(4.03) 2.敌敌畏(Dchlorvos)(4.15)
3.速灭戚(Tsumacide)(5.84) 4.氧化乐果(Omethoate)(6.06)
5.仲丁戚(BPMC)(7.38) 6.甲拌磷(Phorate)(8.27)
7.乐果~Dimethoate)(8.7O) 8.呋喃丹(Furadan)(8.81)
9.甲基对硫磷(Parathion-methy1)(10.70) 10.对硫磷(Parathion)(12.10)
方法二:HP-170l,l9091U-102石英毛细管柱(25m×0.20mm×0.20μm);载气为高纯氮气,柱压为140kPa,柱温为90℃,保持0.5min,以25℃/min升至16O℃,再以10℃/min升至240℃;进样口260℃,不分流进样,吹扫时间0.5min,吹扫流量为100mI/min;检测器温度290℃,氢气为3mI/min,空气为60mI/min,尾吹气为10mI/min。
在上述试验条件下,10种农药得到很好分离,分析时间短,注意峰的保留时间及出峰顺序与方法一不同,见图-2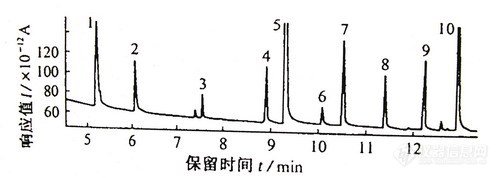
图-2 农药标准色谱图 7550-1
1.敌敌畏(Dichlorvos)(5.19) 2.甲胺磷(Methamidophos)(6.07)
3.速灭威(Tsurrmcide)(7.54) 4.仲丁威(BPMC)(8.90)
5.甲拌磷(Phorate)(9.30) 6.氧化乐果(Omethoate)(10.08)
7.呋喃丹(Furadan)(10.51) 8.乐果(nmethoate)(11.39)
9.甲基对硫磷(Parathion—methy1)(12.23) l0.对硫磷(Parathion)(12.94)
方法二优于方法一可以将乐果与呋喃丹,敌敌畏与甲胺磷完全分离。
3 结果与讨论
2-1样品液的提取与净化
样品液的提取与净化是试验极为重要的环节,国标方法中提取与净化过程比较复杂,容易引起样品的损失。本法直接以丙酮、二氯甲烷混合液提取,旋转蒸发浓缩,挥干后以丙酮定容即可,直接进色谱分析,过程较为简便,省去了净化步骤,且检测过程中各峰分离及峰形理想,基线稳定,能满足检测需要。
2-2回收率、相对标准偏差及检出限
以苹果、茭白、四季豆、葱为试样(各取10.0g)分别加混合标准使用液1.0mL及5.0mL,进行回收试验。每个平行重复5次,用空白作对照,测定结果,回收率为75.6%~103.7%,RSD为3.0%~8.5%,检出限为2~16μg/kg。
2-3定性与定量
采用保留时间定性,以峰面积外标法定量。由于果蔬提取液成分复杂,使用单一毛细管柱往往会有不明杂质峰干扰,出现假阳性情况,影响测定结果。因不同的成分(包括干扰成分及目标成分)在不同类型的毛细管中的保留时间及出峰顺序不同,本法采用方法一作第一次测定,对有阳性的样品再以方法二复核测定。对仍然为阳性的样品判为检出,否则为干扰。
对送检的25份果蔬样品中,用方法一检出阳性18份,对这18份样品用方法二复核检验后发现3份为杂质干扰,干扰率达16.7%。因此,在缺少质谱定性的情况下,用双毛细管柱分析可大大提高分析的准确性。由此可见,本法检测农药品种多,样品提取简便,分析时问短,准确性高,回收率好,能满足果蔬中残留农药的检测需要
参考文献:
[1] 祖东云.毛细气相色谱法测定多种有机磷农药在蔬菜上的残留[J].中国卫生检验杂志,2000,10(5):570.·27·
-
+关注
私聊
-
yhl-87_
第25楼2009/11/05
农药残留检验方法系列讲座(25)
气相色谱-质谱法测定蔬菜中有机磷农药的残留量
摘要:本文是以GC-MS联用技术检测蔬菜中有机磷农药的残留量,通过谱库检索定性;样品前处理采用了先除样品中水分,再以甲胺磷⑴ (Metamidophos)和马拉硫磷(Malathion)在蔬菜中残留为代表进行提取检测。本文是安徽省教育厅自然科学基金资助项目部分内容,由安徽大学现代实验技术中心的丁毅和安徽大学生命科学学院的倪小东、李飞红、刘晓颖共同完成,具体介绍如下:
蔬菜中有机磷类农药残留测定的国标法为毛细管气相色谱-氮磷检测法(GC-NPD)。虽然以灵敏度高、定量准确、成本低而被广泛采用,但是当遇到组分不明的干扰物与被测物的峰相互重叠,或两者的保留时间非常接近时,则难以判断⑵。气相色谱-质谱法(GC-MS)适合于多残留分析,它能在多种残留物同时存在的情况下对其进行定性定量分析,如只用气相色谱法分析常需要多个检测器,而GC-MS法一般只需一次提取和一次检测,并且可以通过谱库检索实现定性,不需标样也能定性。离子阱质谱不仅可以作一般质谱的工作,还可以实现时间上的多级串联质谱(MSN)。随着GC-MS技术的不断发展和标准谱库的不断扩大,快速准确的检测农药残留成为可能⑶。
1、 实验部分
1-1 仪器与试剂
Saturn2200型GC-MS联用仪(美国Varian公司);WH-3电动振荡器(上海沪西分析仪器制造厂);丙酮(分析纯);无水硫酸钠(分析纯);活性炭:用3mol/L盐酸浸泡过夜,抽滤,洗至中性,在l20℃下烘干备用;农药标准品:甲胺磷、马拉硫磷:99%标准储备液100μg/mL(农业部环境保护科研监制所);标准溶液的配制:将100ug/mL标准储备液临用前丙酮稀释成1μg/mL的标准使用液,贮藏于冰箱中。
1-2 样品的提取
称取青菜试样5.00g,置于研钵中与无水硫酸钠研成干粉状,加入丙酮溶液30mI。震荡提取30min,静置5min,过滤,重复一次;上清液加人少量活性炭去色素后过滤,收集滤液避光浓缩至近干,定容至5mL,进行CC-MS分析。
1-3 气相色谱-质谱条件
色谱条件:选用VarianCp-5860弹性石英毛细管柱(30m×0.25mm),初始温度50℃,以6℃/min升至240℃,运行时间50min,进样口温度200℃,载气高纯氦,用面积归一化法定量。
质谱条件:EI离子源,电离能70eV,扫描测定范围30-650u.离子源温度l50℃,接口温度240℃,电子倍增电压l800V。
2、结果与讨论
2-1 样品前处理方法的选择
由于甲胺磷极性大,水溶性强,易氧化分解,前处理方法采用先用无水硫酸钠与样品研磨至干粉状,使样品中的水分被无水硫酸钠吸于,无水硫酸钠既是吸水剂又是分散剂,研磨后固相颗粒较小,物理上增大了接触面积,提高了提取效率;再用丙酮提取,省去了二次萃取的过程,不仅节约了试剂,而且避免了乳化现象的发生,如图I所示。
GC—NPD检测虽然可以满足灵敏度的要求,但其阳性结果也需要进一步确认,而且对净化要求较高。GC-MS法可以对蔬菜中甲胺磷,马拉硫磷等有机磷杀虫剂同时进行定量测定和阳性确证。样品中常常有干扰物存在,影响对目标化合物的检测,采用MS-MS法,可以选择目标化合物的特征离子做母离子,能从干扰物中检测出目标化合物。
以甲胺磷的分子离子rn/z 141作为母离子,进行MS-MS分析,得到m/z主要为126,111,64碎片离子,根据这些碎片离子可以推断出样品中存在着甲胺磷。在离子检测前就排除本底干扰,即使对于复杂样品也可以达到很高的灵敏度。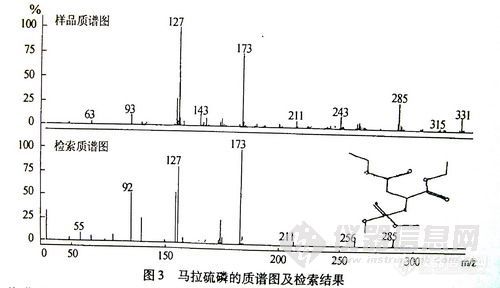
2-3 标准工作曲线的绘制
根据各农药的检测灵敏度,将农药标准储备液用丙酮逐级稀释,配制成6种农药的混合标准工作曲线。按上述色谱条件分析,以峰面积у,对各组分质量х求回归方程见表2。
表2方法的线性范围和检出限 7557-1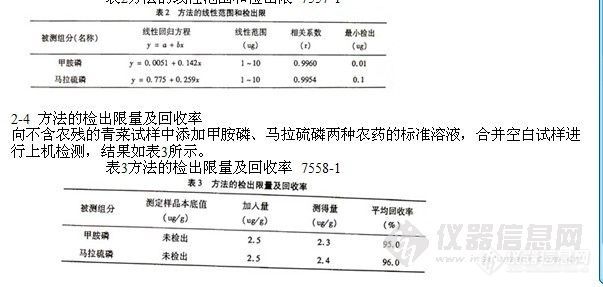
参考文献:
[1] 马建强,等.气相色谱-火焰光度法测定蔬菜甲胺磷残留量 [J].广西师院学报,2001,18(2):6.
[2] 王军,等.农药残留速测技术研究发展 [J].环境污染治理技术与设备,2001,18(1):18.
[3] 刘永波,等.气质联用快速检测蔬菜水果中农药多残留的方法 [J].青岛科技大学学报,2003,24(6):492
-
+关注
私聊
-
yhl-87_
第26楼2009/11/05
农药残留检验方法系列讲座(26)
一种新的农药残毒快速检测装置
摘要: 本文介绍一种新的农药残毒快速检测装置。该装置基于酶催化动力学原理,以酶抑制率反映蔬菜中残留农药的总毒性。该装置集化学分析、光机电、信息、生物等技术于一体,可对六种样品进行同时检测。测量准确度高,精密度好,方便快速,适合于各种实际应用场合,具有广泛的应用前景。
1 前言
农药是农林牧业发展的保障,同时也是环境和食品的重要污染源之一,农药残留问题越来越受到各国政府和公众的关注。寻求一种快速准确的分析检测方法已成为一件迫在眉睫的事情。目前,农药中毒事件多由有机磷和氨基甲酸酯类农药引起,针对这些农药残留的分析方法以色谱法居多①~④,但操作繁琐、费时。有关农药残留速测仪的研制成果⑤,均未考虑到温度对酶催化动力学反应的影响,且所配套的酶试剂包不适应现场操作需要,实际应用效果不够理想。而基于酶抑制法的试纸目视比色法虽方便快捷⑤,但其灵敏度和准确性差。
本文的介绍是由吉林大学化学学院的杨蕊、郑健、于爱民和金钦汉;中国检验检疫科学研究所的邹明强和储晓刚;吉林出入境检验检疫局的牟峻等研究人员共同在以前研制的手持式农药残毒速测仪⑦的基础上,新研制了一种六通道农药残毒速测仪,基本上克服了上述缺点,可实现多种样品同时快速检测,其准确度和灵敏度也令人满意。现全文介绍如下:
2 实验部分
2-1 仪器及试剂
自制六通道农药残毒速测仪:超声提取器由昆山市超声仪器有限公司生产的KQll8型超声清洗器改装而成。
缓冲溶液:0.1mol/L的磷酸盐水溶液,pH为8.02;
底物溶液:适量碘化硫代乙酰胆碱(acetylthio-choline,ATchI)用水溶解;
显色剂溶液:缓冲溶液溶解适量的二硫代双硝基苯甲酸(DTNB);
酶溶液:缓冲溶液溶解适量乙酰胆碱酯酶(Acetylcholinesterase,AchE)。
2-2 检测方法
测空白: 于比色瓶中加人4mL磷酸盐缓冲溶液⑤,放入仪器中调零,加入80μL酶和80μL显色剂,放入仪器中在37℃培养l0min,加入80μL底物,振荡均匀,放回仪器中按下测定键,仪器会自动测当时的吸光度以及三分钟后的吸光度,并计算出差值。
测样品: 称取1.0g蔬菜样品,剪成1cm见方大小,放人样品瓶中,再向其中加入l0mL缓冲溶液,超声提取3min,室温放置10min,然后加入酶和显色剂各80μL,以后各步骤同“测空白”。仪器可以根据前面测得的四个吸光度值自动计算出酶抑制率,并显示在液晶屏幕上。
2-3 检测装置结构与性能
仪器具有六通道同时检测功能,包含有六套检测装置,每套均有LED光源、样品池、检测器,各成体系,无可动部件。六套装置由单片机控制,步调一致,自动计时、计算,提高了准确度和灵敏度。
2-3-1 农药残毒快速检测仪结构设计 仪器结构如图1所示。 7914-1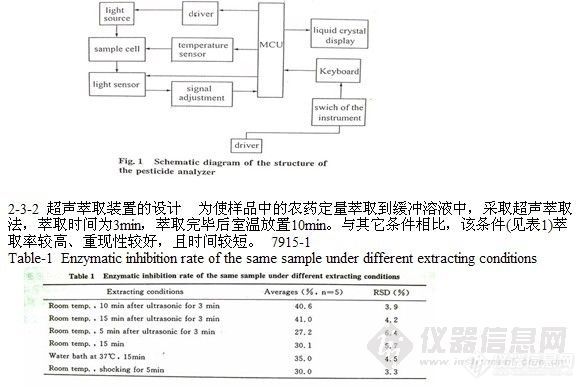
2-3-3 试剂包装的设计 乙酰胆碱酯酶是一种室温下不稳定的物质,需要低温保存。我们采取液体酶加酶稳定剂技术,将各种试剂配成溶液,装人小玻璃安瓶中,抽真空后密封,从而可长时间保存(室温下可保存3个月,冷藏可保存一年以上),可满足实际现场检测需要。2-3-4 技术指标
光源:超高亮度发光二极管;
波长:410±2nm;
吸光度范围:0~2.0;
吸光度相对标准偏差(RSI)):±39.6;
抑制率范围:0~100%;
检出限:0. 1~3.0mg/kg (以抑制率25%计);
电源:220V±l0%,50Hz;
重量:5kg
尺寸:390×260×200mm。
3 结果与讨论
3-1 六通道平行性实验
六通道同时对同一样品进行抑制率测定,相对标准偏差不超过4%。
3-2 试剂滴加方法考察
为使操作方便,采用特定的滴瓶加入各种试剂。经测定,每滴试剂均为40μL,每次加入各种试剂均为两滴。采用此滴瓶加入试剂与使用移液器对同一样品测定结果相比较,相对误差不超过2%。
3-3 样品预处理条件
目前测定蔬菜样品农药残留的前处理多采用直接有机溶剂提取⑨⑩或有机溶剂提取后再用柱层析净化⑾,这些方法操作复杂、费时,而且要采用大量有毒溶剂,本法只需将样品剪成1cm见方的小块,然后用磷酸盐缓冲溶液超声提取,因而具有方便、省时、无毒、无环境污染的优点。另外,仪器在0~2.0吸光度范围内线性范围良好,因此足以使样品测量点所对应的吸光度值扣除本底值 (一般吸光度值小于0.7) 后仍在线性范围,从而蔬菜样品中的基体干扰对测量结果几乎没有影响。
3-4 测量准确度和精密度
对添加农药的相同蔬菜样品进行农药残留测定,并以气相色谱法作对照实验,其检测结果相符,如表2所示。
3-5 实际应用
使用本仪器对实际样品进行了测定,结果如表3所示,其中抑制率超过25%为可能含有农药残毒,超过40%为一定有农药残毒。
Table 2 Comparison of concentration obtained by using this method and traditional method from commercial production 7916-1
2-6 特点及应用前景
仪器设计了模块化技术的六套全固态光源和检测器,具有温度控制功能,使酶催化动力学显色反应在恒温下进行,克服了温度对化学反应速度的影响,保证了测量结果的重现性,可实现六通道同时测量,分析速度快(每小时可测定120个样品);采用双重保护技术使酶试剂在0~4℃下保质期为1年以上,在室温下可稳定三个月以上;有判断操作错误和试剂是否失效功能,样品的培养、生化和化学反应、显色和测定一体化,采用超声波萃取技术,提取完全、操作简便。基于以上特点,本仪器可应用于蔬菜农药残毒的现场快速检测,具有相当广泛的应用前景。
参考文献:
1. Eliezer Zomer,Steven Saul,Stanley ECharm.BiotechnologyAdvances [J ],1995,13 (4):763.
2. Rastrelli L,Totaro K,De Simone F.Food Chemistry [J ],2002,79 (3):303.
3. Russo M V ,Campanella L, Avino P . Journal of Chromatography B-analytical Technologies in the Biomedical and Life Sciences [ j ] ,2002,780 (2) :431
4. Galloway T S,Millward N,Browne M A.Aquatic Toxicology [J],2002,61 (3-4):169.
5. 黄文风,蔡琪,黄敏.分析测试学报 [J ],2000,19 (6):87.
6. 杨大进,中国食品卫生杂志 [J ],1998,10 (2):38.
7. 邹明强,杨蕊,于爱民,金钦汉.高等学校化学学报 [J ],2003,24 (6),1016.
-
+关注
私聊
-
yhl-87_
第27楼2009/11/05
农药残留检验方法系列讲座(27)
气相色谱-质谱法研究
敌百虫在气相色谱分析过程中产生的分解产物
摘要:
本文由农业部上海水产品质量监督检验测试中心的于慧娟,蔡友琼,李庆,毕士川,黄冬梅等研究人员,利用气相色谱-质谱联用技术对敌百虫在气相色谱分析过程中产生的分解产物进行定性研究,有利在实践分析中区别是敌百虫还是敌百虫的分解产物,并在分析操作中加以注意。通过质谱解析鉴定出敌百虫的分解产物有三氯乙醛、磷酸二甲酯和敌敌畏。选择了气相色谱条件,如进样口温度、柱温升温速率、进样方式对敌百虫分解产物的影响。实验结果表明,这3种因素对敌百虫的稳定性均有不同程度的影响,其中进样口温度是导致敌百虫分解的主要因素,进样口温度越高,敌百虫的分解量就越大。
1 前言
敌百虫是一种有机磷酯类杀虫剂,由于具有良好的驱虫效果而被广泛地用于农副产品的种植业、养殖业和加工业中。由于敌百虫具有一定的毒性,长期食用含有敌百虫残留的食品会对人体产生不利的影响,因此准确地测定出食品中敌百虫的残留量就显得十分重要。
敌百虫的分析方法很多,有容量法⑴、分光光度法⑵、气相色谱法⑶~⑻和液相色谱法⑼~⑾。在食品和植物中有关微量敌百虫残留量的测定大多采用气相色谱法,所用检测器有电子捕获检测器(ECD)、氮磷检测器(NPD)和火焰光度检测器(FPD)、氢火焰离子化检测器(FID)。这地方法虽然灵敏度高,重现性好,但不适合于敌百虫残留量的分析,其原因是敌百虫热稳定性差,在气相色谱分析过程中很容易发生分解⑽⑾,且有多种分解产物,严重影响了敌百虫定量的准确性。有关敌百虫在气相色谱分析过程中所产生的分解产物的种类及量国内外尚未见报道。本文利用气相色谱-质谱联用(GC/MS)技术对敌百虫在分析过程中所产生的分解产物进行了定性与定量研究,通过质谱解析确定了分解产物的种类和结构,为选择最佳的敌百虫分析方法提供了可靠的实验依据。
2 实验部分
1-1 仪器和试剂
Agilent6890N/Agilent5973i气相色谱/质谱联用仪,配有7683型自动进样器。丙酮(农残级,Sigma公司);
敌百虫和敌敌畏 (纯度≥99%,上海农药检定所);
磷酸二甲酯和三氯乙醛 (纯度≥99%,Sigma公司)。
敌百虫标准溶液:用丙酮将敌百虫配制成质量浓度为11mg/L的标准溶液。
1-2 分析条件
色谱条件: 色谱柱HP-5MS毛细管柱(30m×0.25mmi×0.25μm);进样口温度:200℃;载气:高纯氦气,流量1.5mL/min;色谱柱起始温度45℃,保持3min,以10℃/min的速率升至250℃,保持3min。脉冲不分流进样,进样量1μL。
质谱条件: 电离方式电子轰击离子源(EI);电子能量:70eV;离子源温度:230℃;接口温度:230℃;扫描范围(m/Z) 10~270;电子倍增器电压为859V;阈值50,溶剂延迟时间2.2min。
3 结果与讨论
2-1 敌百虫分解产物的定性研究
为了解敌百虫的分解状况,作者利用GC/MS技术按“1-2”节条件对敌百虫的标准溶液进行了分析,并对分析过程中出现的每一个色谱峰相应的质谱图进行人工解析和计算机检索 (NIST谱库),确定其化学结构,经标准品进一步确认后,最终发现敌百虫在气相色谱分析过程中同时有3种分解产物产生,这3种分解产物是三氯乙醛、磷酸二甲酯和敌敌畏。敌百虫的总离子流色谱图及分解产物的质谱图见图1~4。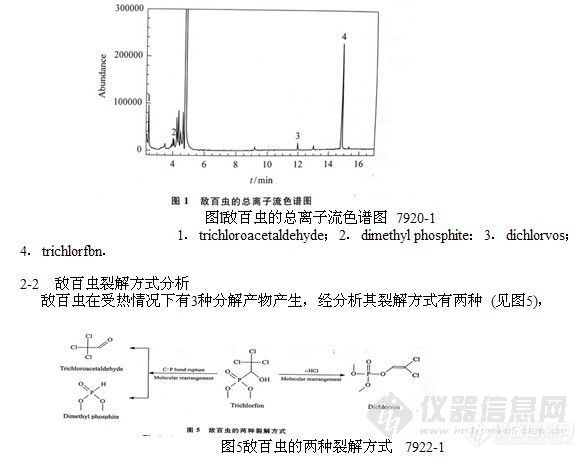
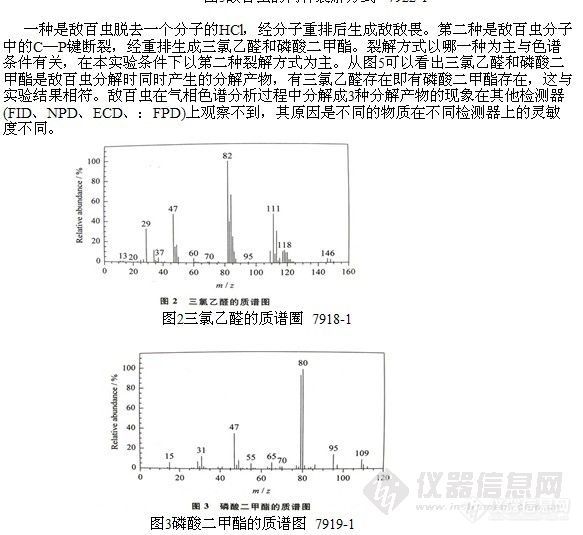
-
+关注
私聊
-
yhl-87_
第28楼2009/11/05
农药残留检验方法系列讲座(27) (下) 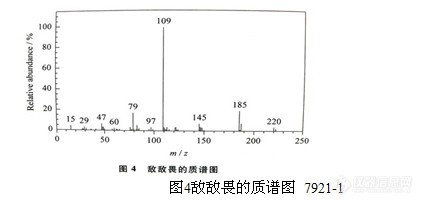
2-3 敌百虫分解的影响因素考察
敌百虫在气相色谱分析过程中分解产物量的大小主要与色谱条件有关。本实验研究了进样口温度、柱温及进样方式 (不分流进样和脉冲不分流进样)对敌百虫稳定性的影响。为减少进样口活性对敌百虫稳定性的影响,本实验中的进样口使用新的去活玻璃衬管。实验发现进样口温度、柱温及进样方式这3种因素对敌百虫的稳定性均有不同程度的影响,其中进样口温度是导致敌百虫分解的主要因素。实验证明进样口温度越低、柱温越低和进样速度越快,敌百虫的分解量就越小。进样口温度对敌百虫稳定性的影响见图6。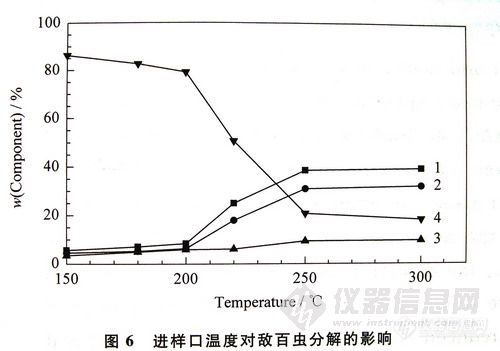
图6进样口温度对敌百虫分解的影响 7923-1
1. trichloroacetaldehyde:2.dimethyl phosphite:3.dichlorvos:4.trichlorfon
由图6可以看出,随着进样口温度的升高,敌百虫的量逐渐减少,而分解产物的量逐渐增大,其中在200~250℃时敌百虫分解迅速,其含量从80%下降到20%。低的进样口温度(≤180℃)虽能在很大程度上降低敌百虫的分解量,但不具有实用价值。这是因为我们分析的对象多为基质复杂的样品,其中含有很多沸点很高的物质,如果进样口的温度设置太低,有些物质不能气化,将导致进样口内的衬管易被污染,分析的重现性差并经常有鬼峰出现。
2-4 敌百虫的定量方法探讨
目前敌百虫的定量方法有两种:一种是测定分解产物,将敌百虫定量地转化为三氯乙醛进行定量,另一种是测定敌百虫母体。从目前的研究结果和我国农药的使用现状来看两种方法都存在着弊端。这是因为利用气相色谱法测定敌百虫母体时,由于敌百虫分解使测定结果不准确或根本检测不到敌百虫,造成定量结果偏低。以分解产物为依据对敌百虫进行定量,只要分解产物稳定,其分解量同敌百虫的量成正比,该方法原则上是可行的,但要根据不同的分析对象选择不同的分解产物,不能一概而论。因为在食品的农药残留分析中,一些分析对象 (蔬菜、粮食、水产品及一些加工食品) 经常会含有微量敌敌畏或三氯乙醛,这就给敌百虫的准确定量带来了问题。因此,要想准确地对微量的敌百虫进行定量,必须根据不同的分析对象来选择不同的分解产物或开发新的检测方法,如冷柱头进样-气相色谱法或高效液相色谱法,以确保测定结果的准确性。
参考文献:
[1] GB334-2001
[2] GBl1893-89
[3] Yang Zuying.Food Analysis.Beijing:Chemical Industry Press (杨祖英.食品检验.北京:化学工业出版社),2001 ,144
[4] Xi Shaofeng.Standard &Quality of Light Industry (席绍峰.轻工标准与质量),
2002,(5):5O
[5] Chen Zhidong,Liu Shuaigang,Chen Ping.:Pesticide Science and Administration (陈智东,刘帅刚,陈平.农药科学与管理),2000,21(1):9
[6] Zhao Lan,Fan Defang.Pesticides (赵岚,樊德方. 农药),1996,35 (9):26
[7] Xie Baomin.Chemical Industry Standard Measurement Quality (谢宝民.化工标准·计量·质量),2003,(11):2l
[8] Yin Derong,Zhou Xiaoping,Wang Liyuan.Chinese。Journal of Health Laboratory Technology (殷德荣,周晓萍,王立媛.中国卫生检验杂志),2005,15 (2):l85
[9] Ma Yaguang,:Xing Jun,Mei Baogui,Wang Baojie.Chinese Journal of Pesticides (马亚光,邢君,梅宝贵,王宝杰. 农药),2002,4l (2):l7
[10] Zhao Ying,Liu Yinggui,Ya Ping,Zhang Ping,Zhao Guiying.
Chinese Journal of Veterinary Drug (赵英,刘迎贵,亚萍,张平,赵桂英. 中国兽药杂志),2005,39 (3):23
[11] Zhang Mingguo,Zou Hongxiang, Tan Ni.Hubei Chemical Industry (张明国,邹鸿享,覃尼. 湖北化工),1999,(5):32
-
+关注
私聊
-
yhl-87_
第29楼2009/11/05
农药残留检验方法系列讲座(28)
中药材柴胡中8种有机磷农药残留测定
摘要 目的:建立同时测定中药材柴胡中8种有机磷农药毛细管气相色谱测定方法。
方法:样品前处理方法简单,样品中加人少量活性炭,可用乙腈直接提取。选用火焰光度检测器(FPD)和DB-17毛细管柱,程序升温进行气相色谱测定。
结果:8种有机磷农药(敌敌畏、甲胺磷、乙酰甲胺磷、氧乐果、久效磷、甲基对硫磷、毒死蝉、喹硫磷)的最小检出量在0.05ng~0.15ng之间,平均回收率在82.07%~107.75%之间,RSD在0.85%~14.00%之间。
结论:该法灵敏度高、选择性强、操作简便、重现性好。
本文由中国农业大学理学院的王素利与刘丰茂和中国医学科学院中国协和医科大学药用植物研究所的陈建民与薛健等研究人员共同完成,现全文介绍如下。
前言
柴胡现以伞形科多年生草本植物柴胡(北柴胡)Buplerum chinese DC的根及狭叶柴胡(南柴胡)B.scorzonerifolium Willd..的根入药。前者生产于辽宁、甘肃、河北、河南、山东等地,后者主要产于湖北、江苏、安徽、四川等地。柴胡具有解表和里、升阳、疏肝解郁等功能。主要用于治疗感冒、上呼吸道感染、疟疾、寒热往来、胁痛、肝炎、胆道感染、胆囊炎、月经不调、脱肛等[1]。人工栽培的中草药在生长的过程中,农药的使用也十分频繁,因此中草药的农药残留不容忽视,它直接关系到患者的身体健康。有机磷农药不仅使用于水果、蔬菜,同样也使用于中草药。有机磷农药在水果、蔬菜中多残留方法已经成熟[2~4],而中药由于成分复杂,现还没有成熟的方法。关于中草药中有机磷农药的多残留分析方法也有一些报道[5~7]。前处理大部分要经过层析柱或固相萃取净化。本文选用常用中药材柴胡对其进行有机磷农药残留方法测定,采用提取和净化同步完成,操作简便,而且方法的回收率、准确度、精密度均符合多残留分析要求。
实验
1 仪器
Agilent 6890气相色谱仪,配备火焰光度检测器(FPD);DB -17弹性石英毛细管柱(30 mm×0.25mm×0.25 μm);空气浴振荡器(HZQ - C);旋转蒸发仪(N -1)(TOKYO RIKAKIKAI CD,LTD)。
2 试剂与药品
醋酸乙酯、乙腈均为分析纯;活性炭(用酸处理调至中性烘干);有机磷农药标准品:敌敌畏(99.0%),甲胺磷(99.0%),乙酰甲胺磷(99.2%),氧乐果(98.9%),久效磷(99.2%),甲基对硫磷(99.8%),毒死蜱(99.0%),喹硫磷(99.5%),由中国农业部农产品质量检测中心提供;中药材柴胡,由中国医学科学院药用植物研究所提供,并经本所潘瑞乐副研究员鉴定。样品名称及来源见表4。
3 供试样品的提取和净化
精确称取过40目筛粉末4.0 g于150 mL具塞三角瓶中,加人乙腈50 mL浸泡,然后加人0.2 g活性炭,在振荡器上振荡提取1 h,滤过,收集25 mL滤液于浓缩瓶中在40℃浓缩近干,吹干后用醋酸乙酯定容1 mL,待检测。
4 色谱条件
进样口温度200℃;检测器温度250℃;柱升温程序:初始温度120℃,保持1 min,以10℃/min程序升温至200℃,以3℃/min升至220℃,再以20℃/min升至240℃,保持3 min;载气流速为(N,):30mL/min;氢气:15 mL/min;空气:100 mL/min。采用不分流进样,外标法定量,进样体积:1μL。
5 线性关系的考察
分别精密称取8种有机磷农药标准品(精确到0.0001 g),用醋酸乙酯配成一定浓度的标准溶液,然后分别取一定量配成8种有机磷农药的混合标准母液(敌敌畏为1.5 mg/L,乙酰甲胺磷、氧乐果为4 mg/L,甲胺磷、久效磷、甲基对硫磷、毒死蜱、喹硫磷为2 mg/L),用醋酸乙酯梯度稀释母液得到1,2,5,20,50倍稀释混合标准溶液,用气相色谱进行分析。标准样品色谱图及8种有机磷农药的出峰时间如图1。以峰面积对农药质量浓度C(mg/L)做线性回归得到的各种农药的线性回归方程见表1。8种有机磷农药在0.03~2 mg/L范围内线性良好,其最小检出量(LOD)和最小检出浓度(根据3倍信噪比确定的)及定量限(L0Q)见表1。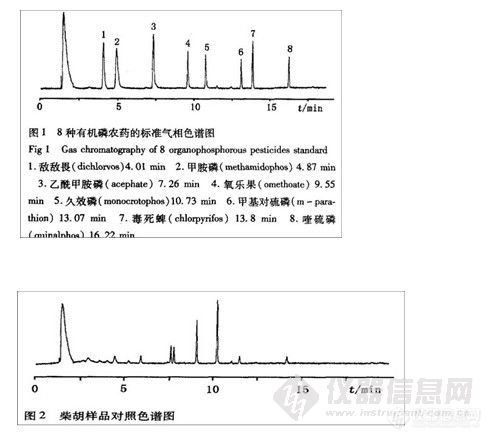
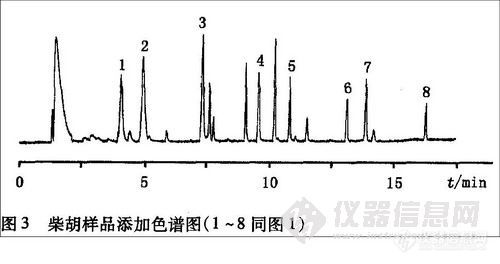

7 回收率试验
在4.0克粉碎样品粉末中分别加人标准工作液0.5,1,2 mL,进行低、中、高3个水平的添加回收试验,同时对其进行空白对照试验,每个水平的添加重复5次。空白对照和样品添加回收图谱如图2图3。 回收率和RSD见表3。 3个添加浓度分别为敌敌畏0.0375、0.075和0.15 mg/kg;甲胺磷、久效磷、甲基对硫磷、毒死蜱、喹硫磷为0.05、0.1和0.2 mg/kg;乙酰甲胺磷、氧乐果为0.1、0.2和0.4 mg/kg。从表3可知本方法的平均回收率为82.1%~107.8%,相对标准偏差为0.9%~14.0%,完全满足农药残留分析的要求。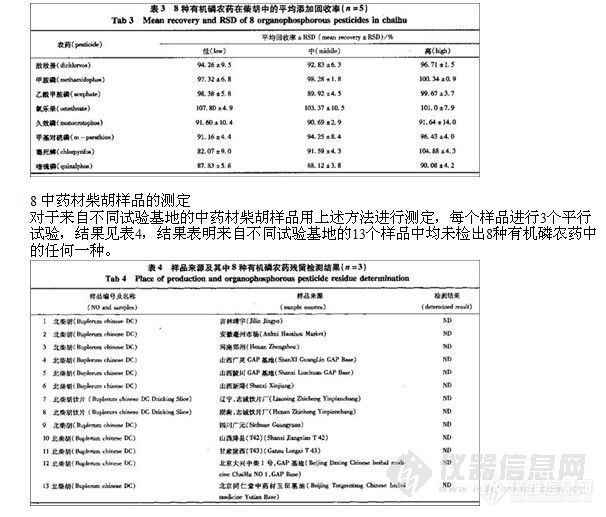
9 讨论
样品前处理方法的选择,作者曾采用丙酮、醋酸乙酯以及二氯甲烷、醋酸乙酯一石油醚、丙酮-石油醚等不同比例提取,经浓缩后直接进样,或过层析柱后进样。结果表明本文采用的方法简便、杂质少,而且方法回收率、准确度、精确度均符合农药多残留分析要求,为中草药中农药多残留快速检测提供了一种较好的方法。
参考文献
1 Chinese herbal medicine compilation group(全国中草药汇编编写组编)·Chinese herbal medicine compilation(全国中草药汇编).People’s Medical Publishing House(人民卫生出版社),1983.5
2 LIU Gang(刘刚),HUANC Rong-mao(黄荣茂),JING Lin-bong(金林红),et al.
Determination of organophosphorus pesticide residues in vegetables(蔬菜中有机磷农药多残留检测方法).Acta Untr Gui zhou(Natural Sci)贵州大学学报(自然科学版),2004,2:68
3 YANG Da -jin(杨大进),FANG Cong-rong(方从容),ZHANGYing(张莹)Determination of organophosphorus and carbamate pesticide residues in vegetables(蔬菜中有机磷和氨基甲酸酯农药多残留的测定).Chin Food Health J(中国食品卫生杂志),1997,9(5):9
4 National Standard of Republic of China(中华人民共和国国家标准)CB/T[S].5009.20 -1996
5 FENG Xiu -qiong(冯秀琼),TANG Qing-yong(唐庆勇).Multiresidue determination of organophosphorus pesticide in Chinese medicinal herbs(中草药中有机磷农药多残留的同时测定)Pesticide Sci Administr(农药科学与管理),2002,23(2):17
6 ZHANG Shu-ming(张曙明),TIAN Jin-gai(田金改),GAO Tian-bing(高天兵).Determination of organophosphorus pesticide residues in medicinal plants and traditional chinese preparations by capi1lary gas chromatography(中药中有机磷农药残留量的毛细管气相
色谱测定方法).J Instrum Anal(分析测试学报),1999,18(5):15
7 LOU Jian(楼健),JIA La-la(加拉拉).Capillary gas chromatography for determining the residues of multiple organophosphorus pesticide in Radix trichosanthis(毛细管气相色谱分析有机磷农药在中药材天花粉中的多残留).Chin J Pharm Anal(药物分析杂志),2003,23:61
-
+关注
私聊
-
yhl-87_
第30楼2009/11/05
农药残留检验方法系列讲座(29)-中药 柱后衍生法测残留
HP$$lc柱后衍生法测定中药材中氨基甲酸酯类杀虫剂残留
摘要 目的:采用高效液相色谱柱后衍生法测定中药材中氨基甲酸酯类杀虫剂残留。
方法:通过乙腈提取,固相萃取(SPE)净化,Waters(3.9m m×150mm×4μm)柱,甲醇一水一乙腈(16:68:16)为流动相,柱温30℃,流速1.5 mL·min-1进行分离,柱后衍生反应系统,OPA和氢氧化钠流速均为0.5 mL·min-1,反应温度80℃,2475荧光检测器进行监测,进样20 μL。
结果:速灭威、克百威和西维因的添加回收率分别为84.6%~110.5%,86.9%~99.8%,90.1%~110.5%;RSD分别为4.0%~8.6%,3.8%~8.5%,3.1~7.5%;最低检测限分别为0.01,0.01,0.005μg·mL-1。
结论:该方法分离效果好,灵敏度高。
本文由农业部农产品质量监督检验测试中心云南省农业科学院的刘宏程和黎其万;云南省烟草研究院的杨光宇;昆明市农产品质量监测检验中心的马雪涛三个单位协作完成的高效液相色谱柱后衍生测定法,现全文介绍如下。
前言
速灭威(tsumacide)、克百威(carbofuran)和西维因(carbaryl)(图1)都是氨基甲酸酯类杀虫剂,属广谱性的杀虫、杀螨剂,具有触杀、内吸作用;近年来在中药材栽培中也大量使用,主要施用于土壤中,防治土壤中的多种害虫和线虫,容易导致在药材中的残留,其残留的存在不仅制约着许多名贵药材进人国际市场,而且对中成药生产也带来新的质量问题。氨基甲酸酯类农药在农作物中残留量的检测已有较多报道[1] [2],而有关中药材中残留量的检测少有报道[3] [4]。随着中药现代化进程的加快,中药材生产质量管理规范(GAP)种植基地的建设迫切需要建立一套残留检测方法,为制定安全使用准则或进行安全性评价提供技术手段。
由于氨基甲酸酯类化合物在高温下不稳定和缺乏灵敏度高的检测器,常用高效液相色谱柱后衍生法进行测定。文献[5] 报道了中药材中涕灭威、克百威和甲萘威残留的反相高效液相色谱紫外检测法,但其样品前处理过程复杂,紫外检测灵敏度低。本文以乙腈为提取溶剂,采用固相萃取(SPE)与柱后衍生-荧光检测的高效液相色谱法,建立了中药材中速灭威、克百威和西维因残留的测定方法。回收率在84.6%~110.5%,RSD为3.1%~8.6%,最低检测限为0.01μg·mL-1。能满足食品检验的需要。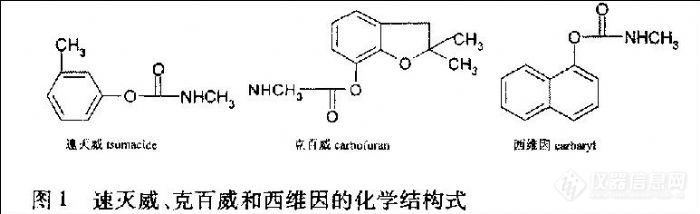
1 仪器及试剂
Waters Alliance 2695高效液相色谱仪,配置四元泵溶剂淋洗系统,自动进样系统,2475荧光检测器,N2吹仪(Organomation Associates Jnc。),固相萃取装置(SUPE$$lc0)及 $$lc - NH2小柱(500 mg,3rnL)(ANPEL),Sartorius纯水器(Germany),高速匀浆机(Heidolph Diax 900)。农药对照品速灭威(tsumacide)、克百威(carbofuran)和西维因(carbaryl)均为100μg·mL-1(农业部环境保护科研监测所)。试剂:邻苯二甲醛(0PA,Acros org,99.5%),巯基乙醇(Farco chemical supplies,99%),氢氧化钠(优级纯),甲醇和乙腈(Fisher,HP$$lc级),乙腈(分析纯),使用前重蒸,四硼酸钠,氯化钠均为分析纯。0.05 moL·L-1硼砂溶液(称取19.1 g四硼酸钠,溶于IL蒸馏水)。0PA溶液(称0PA 50 mg溶于甲醇5 mL,转移到500 mL,容量瓶,用0.05 moL·L-1硼砂溶液定容,过0.45μm滤膜,加人巯基乙醇25μL,摇匀,当天使用当天配制)。0.05 moL·L-1氢氧化钠溶液(称取氢氧化钠1 g,溶于蒸馏水500mL,过0.45μm滤膜)。
样品:茯苓Poria cocos(Schw.)Wolf云南保山;葛根Pweraria lobata(Willd)0hwi云南昆明;何首乌Polygonwm rnultiflorurn Thunb云南昆明。以上中药材均由云南省农业科学院药用植物研究所李晚谊副研究员鉴定。
2 色谱条件
色谱柱是Waters公司专为分离氨基甲酸酯类杀虫剂而生产的色谱柱carbmate analysis(3.9m m×150mm×4μm),流动相:甲醇-水一乙腈(16:68:16),柱温30℃,流速1.5 mL·min-1,柱后衍生系统,0PA溶液和氢氧化钠溶液流速为0.5 mL·min-1,反应温度80℃,2475荧光检测器,激发波长和发射波长分别为339 nm,445 nm;进样20μL。
3 线性关系
分别取1 mL速灭威、克百威和西维因标准溶液(100μg·mL-1),混合,用甲醇定容至10 mL容量瓶,再分别稀释为0.05,0.1,0.25,0.5μg·mL-1的混合溶液,取20μL进样,测定峰面积,以峰面积对浓度作曲线。
速灭威、克百威和西维因在0.05~0.5μg·mL-1的浓度范围内线性关系良好,在此范围内定量准确。在所选条件下,速灭威、克百威、西维因的回归方程分别为:
C = 72.l071X-114.1101 r =0.9979
C = 148.4131X-314.4853 r =0.9979
C = 83.5351X -122.2998 r =0,9979
检出限分别为0.01,0.01和0.005μg·mL-1,标准谱图见图2(第一个峰是速灭威TR=4.393,第二个峰是克百威TR=4.985,第三峰是西维因TR=5.400,最后一个峰是抗蚜威TR=6.036);此色谱条件可同时分离抗蚜威农药残留,由于抗蚜威是氨基甲酸二酯,碱水解不产生甲酯,不能进行衍生化反应,在此波长下灵敏度不高,但抗蚜威自身带有荧光,通过时间编程控制其波长变化可提高抗蚜威的灵敏度,图中第四个峰(抗蚜威)的基线上移正是由于此段时间内波长改变所引起的。
4 精密度和回收率实验
取速灭威、克百威和西维因1.0μg·mL-1的混合对照品溶液20μ L,连续进样5次,测得RSD分别为1.72%,2.13%和2.16%。取茯苓、何首乌和葛根3份,分别加人速灭威、克百威和西维因混合溶液1·0μg·L-1和2.5μg·mL-1各1 rnL,进行回收率实验,计算平均回收率,精密度和回收率见表1。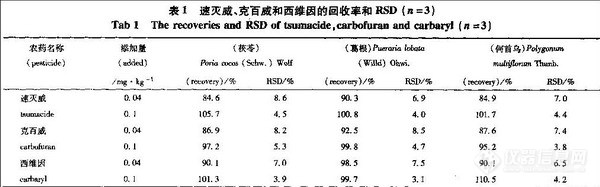
5 样品溶液制备及测定
准确称取25 g粉碎样品于250 mL锥型瓶中,加水10 mL浸泡,振动3 h,加乙腈50 mL,在高速匀浆上匀浆2 min,将匀浆好的样品过滤到100 mL的具塞量筒(量筒内预先放4 g氯化钠),收集滤液后盖上塞子,激烈振荡1 min,静置10 min,待乙腈和水完全分离。准确吸取l0 mL有机层于50 mL烧杯置80℃蒸汽上用N2吹至近干,取出再用空气轻吹赶除残留的溶剂。取$$lc- NH2小柱,先用甲醇:二氯甲烷(1:99)5 mL冲洗柱子,再用甲醇:二氯甲烷(1:99)2 mL溶解样品,上柱,控制流速在2 mL·min-1,收集洗脱液于10 mL离心管中,甲醇:二氯甲烷(1:99)再洗二次柱子每次2 mL,真空抽干,N2吹仪上吹干,用甲醇准确定容至2.0 mL刻度离心管中,经0.45μm滤膜过滤,取20μL进样,测定峰面积,按外标法定量。
6 结果与讨论
6·1 样品的提取
由于中药材的制作方法都是晒干,水分很少,需要加人少量的水来分散颗粒的表面,使它包裹着的农药分离出来,如果不用水浸泡,有机溶剂直接提取,则影响速灭威的回收率,低于70%。加人的水可在过滤的时候可通过盐析干燥。
6·2 溶剂的浓缩
乙腈萃取液是通过N2吹仪进行浓缩至近干,此过程一定要注意,如果在N2吹仪溶剂完全吹干了,则农药会被空气氧化,回收率比较低;如果有乙腈残留,则会影响下一步固相萃取的分离效果。此步正确的操作是N2吹至烧杯还剩1~2滴液体,取出再用空气轻吹赶除残留的溶剂,然后加人甲醇:二氯甲烷(1:99)溶解1 mL,以备净化用。
6·3 优缺点
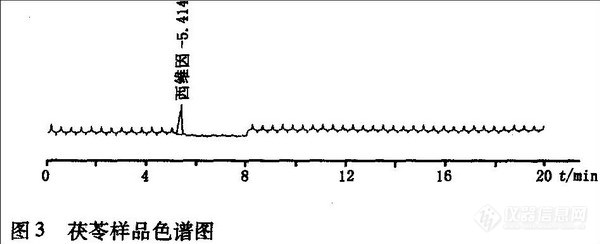
荧光检测器只对带荧光基团的物质有响应并且比紫外检测器灵敏,因此通过柱后衍生化反应可有效去除杂质对样品的干扰,从样品谱图上可看出,几乎没有其他杂质的干扰,可大大提高氨基甲酸酯类农药残留的检出灵敏度。但是,需要在高效液相色谱的基础上增加柱后衍生反应装置,需要增加额外的投人,而且此色谱柱专用性强,主要用于氨基甲酸酯类杀虫剂的分离。对市售中药茯苓、当归、砂仁、黄芪等多个品种进行测定,均取得良好效果,其中茯苓检测出西维因农药残留,如图3。其它样品均未检出氨基甲酸酯类农药残留。