-
+关注
私聊
-

yhl-87_
第41楼2009/11/06
农药残留检验方法系列讲座(36)(下)-柱后光化学反应-HPLC-茶叶中农药
3. 4 光化学反应的时间及PTFE 管长度的选择
在PTFE 管长度一定时,光化学反应的时间随着流动相流速的加快而缩短,荧光信号也随着流速的加快而变小。图3 为PTFE 管长度为15m 时流动相流速对荧光信号强度的影响。综合考虑荧光信号强度、谱峰变宽和色谱分离效率,确定1.0 mL /min 为流动相的最佳流速。此外,还进行了在一定流速(1.0 mL /min)下,实验了不同长度(分别为6m 、10 m 和15 m )的PTFE 管对荧光信号的影响,结果表明:在管长为15 m 时的荧光信号最大。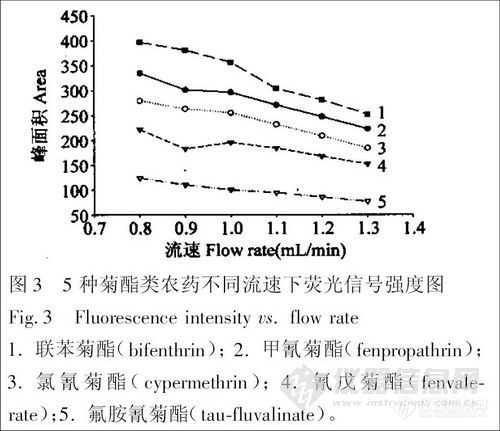
3. 5 光化学反应温度
光化学反应的温度对光化学反应的速率与效率有重要的影响。实验采用能精确控制温度的恒温箱来调节光化学反应的温度,研究了不同反应温度对荧光信号强度的影响(如图4)。图中的结果表明,甲氰菊酯、氯氰菊酯、氰戊菊酯、氟胺氰菊酯在温度为68℃ 时荧光信号(峰面积)最大,联苯菊酯的荧光信号(峰面积)随着温度的升高而增大,综合考虑各组分的荧光信号强度,本实验采用的光化学反应的温度为68℃ 。
3. 6 方法的检出限、线性范围和精密度
实验采用峰面积的方法对待测目标物进行定量。表1 列出了方法的检出限、线性范围以及相对标准偏差。检出限定义为信噪比(S/N)为3 时所对应的方法检出限[17],其中噪音测量的时间为20 min 。表1 中的检出限已经换算为固体茶叶样品中最终能检出的目标物的含量(μg/g),即样品制备中称取的茶叶样品为1.000 g,经提取、净化、浓缩后的测定溶液体积为0.20 mL 。表中列出的线性范围是工作中实际验证的目标物含量范围。在此浓度范围内,待测组分浓度与其相应的色谱峰面积呈线性关系,对超出该范围的较低或较高浓度,并未进一步验证。相对标准偏差是0.1 mg /L标准溶液平行进样8 次实验所得。
3. 7 实际样品测定
实际样品测定时按文献[15 ]的方法 对样品进行预处理,按优化后的实验条件进行测定。4 个茶叶样品的测定结果见表2。方法的检测能力完全满足欧盟对茶叶中农药最高残留量(MRL )新标准的分析要求。方法具有溶剂使用量少、净化效果好、操作简便、快速、选择性好等特点。
-
+关注
私聊
-
yhl-87_
第42楼2009/11/06
农药残留检验方法系列讲座(37)-GC-MS测水中七种氨基甲酸酯残留
气相色谱-质谱法同时测定水中
7种氨基甲酸酯类杀虫剂
前言
氨基甲酸酯类杀虫剂是继有机氯、有机磷之后出现的一类重要杀虫剂.具有作用迅速、选择性高、易分解、残留毒性小等优点,而被广泛使用。但随着此类杀虫剂使用范围的不断扩大.大气、水体、土壤等环境遭到不同程度的污染,由其引起的食物中毒、误服以及投毒事件时有发生.因此有必要建立一种多种氨基甲酸酯类杀虫剂的分析检测方法。为环境检测和临床诊断提供依据。氨基甲酸酯类杀虫剂的测定方法主要有薄层层析法、气相色谱法、液相色谱法⑴ ⑵、液相色谱-质谱法、气相色谱-质谱法⑶-⑻等,但未见GC—MS同时测定该7种氨基甲酸酯类杀虫剂的报道。
本文是由成都疾病预防控制中心的高玲、杨元、王炼及成都市成华区疾病预防控制中心的李仲祥等研究人员共同完成的,他们用气相色谱一质谱法同时测定水中7种氨基甲酸酯类杀虫剂,曾用于井水、自来水、矿泉水、纯净水等水样中微量氨基甲酸酯类杀虫剂的检定,结果满意。下面全文介绍如下:
1、材料与方法
1-1试剂与仪器
氨基甲酸酯类杀虫剂标准溶液:准确称取呋哺丹、西维因、速灭威、残杀威、仲丁威、异丙威、抗蚜威等7种杀虫剂标准品(购自Sigma公司).用甲醇配成标准储备液。置冷暗处密闭保存.临使用时稀释成单一和混合标准使用液;
甲醇:色谱纯;
ENVI—l8小柱:300mg/3mL(美国SUPELCO公司);
固相萃取装置,带采样器(美国SUPEI;CO公司)
GC-MS: HP6890 GC/5973 MSD;
色谱柱:.HP-35MS毛细管柱(30m×0.25mm×0.15μrn)
1-2 实验方法
⑴ GC—MS分析条件
色谱:柱温为80℃(1min)以10℃/min升至240℃再以100℃/min升至270℃;进样口:280℃,脉冲不分流。脉冲压力为45psi。进样量:lμL。载气流速:1.5ml/min,线速度:45cm/sec。
质谱:EI源,电离能量为70eV。传输线为280℃。全扫描(Scan)测定方式的扫描范围为m/z 50~5500选择性离子萃取(SIM)测定方式(扫描离子):速灭威:108;异丙威12l、l50;仲丁威;12l、l36;残杀威ll0;呋喃丹:164;抗蚜威166;西维因144。
⑵ 样品处理
取1000ml水样于烧杯中,将水样和固相萃取装置通过采样器连接,真空泵抽滤,使水样通过ENVI—l8小柱,让待测物吸附在小柱上;然后用甲醇洗脱小柱,定容至1ml,GC/MS测定。
⑶ 测定 按⑴设定分析条件,仪器工作稳定后,直接进样测定。
2、 结果与讨论
2-1 固相萃取条件优化
⑴ 萃取流速和洗脱流速的选择 实验表明,加标水样通过小柱的流速在7.5ml/min以下时,实验可以获得比较满意的回收率;洗脱液通过小柱的流速在0.6ml/min以下时,实验可以获得比较满意的回收率。本文选择水样流速为7.5ml/min。洗脱液流速为0.6rnl/min。
⑵ 洗脱液的体积选择 实验发现,以甲醇洗脱小柱时,分别使用0.8 、0.4、0.4mL甲醇分3段洗脱(每1次均抽至无液体流出时,再补加甲醇),最后定容为1mL实验可获得比较满意的回收率。
2-2 GC-MS条件优化
⑴ 色谱柱的选择
实验发现,中等极性的HP-35MS毛细管柱对7种氨基甲酸酯类杀虫剂能实现基线分离,见图-1
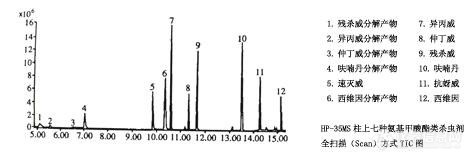
图-1 HP-35MS柱上7种氨基甲酸酯类杀虫剂全扫描方式TIC图 7468-1
⑵ 柱温箱温度优化
氨基甲酸酯类杀虫剂的热稳定性较差,如果色谱柱起始温度太高易发生分解⑤,但起始温度太低,分析时间又过长。经实验考察,本文优化柱温箱温度为80℃ 保持1min以10℃/min升温至240℃,再以100℃/min升温至270℃。
⑶ 进口温度优化
实验表明,当进样口温度为280℃时,所得色谱峰灵敏度最高,色谱峰分离良好。本文选择进样口温度为280℃。
⑷ 进样方式及进样口压力优化
实验表明,当进样口脉冲压力为45psi时所得色谱峰尖锐、灵敏度最高。本文选择进样口脉冲压力为45psi。
⑸ 柱流速优化
实验表明,当载气流速为1.5mL/min时,混合标准的分离效果最好,所得色谱峰灵敏度较高。本文选择载气流速为1.5mL/min。
2-3 质谱检测方式的选择及方法检出限
实验结果证实,在定量分析时SIM检测方式无论在灵敏度、精密度等方面均优于Scan所包含的大量定性信息又是SIM无法与之相比的。因此,本文在定性时采用Scan检测方式,而定量时采用SIM检测方式。结果见表-1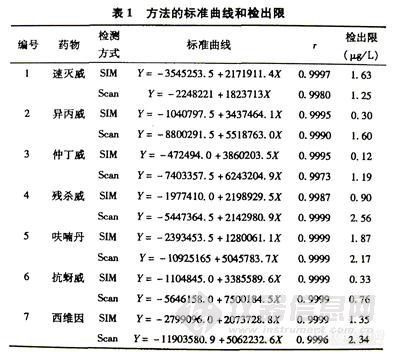
2-4 样品分析
选择不同的水样作为本底,加入7种氨基甲酸酯类杀虫剂混合标准溶液,充分混匀,按前述分析方法进行提取,分别采用全扫描(Scan)测定方式和选择性离子萃取(SIM)方式进行测定,2份加标水平进行5次重复测定,从实验结果看出7种氨基甲酸酯类杀虫剂的回收率分别为:异丙威83.4%~107.7%、仲丁威83.9%~108.5%、残杀威 85.4%~118.0%、呋喃丹82.4%~108.9%、抗蚜威 81.6%~93.9% 、西维因83.6%~94.3%;RSD%在0.55%~10.1%、速灭威为 46.4%~81.9%,符合农药多威留检测的回收率和RSD要求。
参考文献
[1] Bakowski R.S.Gas chromatographic-mass spectrometric confirmation of ten N-methyl earbamati insecticides in liver [J].J AOAC lnt,I994,77(6):1568—1574.
[2] 张洪兰,罗毅.气相色谱法分离检定血中八种氨基甲酸酯 [J].色谱,1994.12(2):117~118,
[3] Yang SS.Recent advances in the residue analysis of N-methyl carbamate pesticides [J].J Chromatogr A,1996,754(1~2):3~16.
[4] Girand D.Determination of traces pesticides in water by solid-phase extraction and liquid chromatography-ionrary mass spectrometri [J].J Chromatogr A,1997,777(1):115—25.
[5] 贾薇,王磊石,赵雷,等.气相色谱-质谱法同时测定七种氨基甲酸酯类杀虫剂 [J].中国卫生检验杂志,2003,13(2):205—206.
[6] Bakowski RS.Gas chromatographic-mass apectrometric confirmation of ten N-methyl carbamate insecticides in liver [J].J AOAC lnt.1994, 77(6 ): 1568~1574
[7] girang D.Determination of traces pesticides in water by solid-phase extraction and liquid chromatography-ionsprary mass spectrometry [J]. J Chromatogr A ,1997,777(1) : 115~154
[8] Climent M J, Miranda MA.Gas chromatographic-mass spectrometric study of photodegradation of carbamate pesticides [J] .J Chromatogr A 1996, 738 (2) : 225~231
-
+关注
私聊
-
yhl-87_
第43楼2009/11/06
农药残留检验方法系列讲座(38)-SPME/SnO2传感器联用快速测果蔬中的有机磷残留
固相微萃取/ 二氧化锡气体传感器联用技术对果蔬中
有机磷农药残留的快速检测
摘 要 研究了固相微萃取(SPME)和二氧化锡气体传感器的联用技术对果蔬中有机磷农药残留乐果、氧乐果、甲胺磷、乙酰甲胺磷、马拉硫磷、敌百虫等的快速检测。结果表明,在85℃ 下,解吸8 min,二氧化锡气体传感器在2 min 内完成对有机磷农药残留的快速检测。零解吸时间测量的甲胺磷的动态响应曲线表明,SPME / 二氧化锡气体传感器联用技术对分析SPME的解吸平衡非常有利。
本文系国家自然科学基金资助项目(No. 602774061)由中国科学技术大学化学系的黄行九、王连超和孙宇峰与中国科学院智能机械研究所的孟凡利和刘锦淮合作完成的部分内容,现将他们完成的论文全文介绍如下
1 引 言
有机磷农药是我国目前生产最多、使用最广的一类农药。研究表明,果蔬中有机磷农药残留严重。有机磷农药是一种神经性毒物,在人体内长期蓄积滞留会引发慢性中毒,导致人体神经功能紊乱,发生出汗、迟钝、神经失常和语言失常等症状。更让人难以始料和防范的是,农药残留在人体内的蓄积,还会通过胚胎和人乳传给下一代,殃及后代的健康[1]。
文献[2 ~ 5]曾报道,利用二氧化锡气体传感器对有机磷农药如敌百虫、乙酰甲胺磷和乐果采用了动态检测原理进行快速检测,获取了大量的反应信息,结合快速傅里叶变换(FFT )频谱分析和极坐标构建,较理想地实现敌百虫、乙酰甲胺磷和乐果的定性和定量检测,大大提高二氧化锡气体传感器对农药气体的选择性和稳定性。
固相微萃取(SPME)是一种基于吸附和解吸的样品前处理技术。近年来,SPME 与其他分析仪器的联用技术成为国内外分析科学家的研究热点,目前对农药检测SPME 主要与gc 、gc / MS 和HPLC 联用[6-14]。本实验将SPME 与SnO2 气体传感器联用,以最常用的有机磷农药如敌百虫、乙酰甲胺磷、乐果、氧乐果、甲胺磷和马拉硫磷为研究对象,结合动态测试方法对果蔬中有机磷类农药残留进行快速检测。
2 实验部分
2. 1 实验方法及样品准备
二氧化锡气体传感器的测试方法采用动态方法,即对传感器采用周期性变化的加热温度,如方波加热,半导体敏感材料的电阻必然随之产生规律性的变化,这个变化依赖于气体的本质特性,而不受外界条件的影响。因此,不同气体和不同浓度同种气体的电阻随温度的变化是不一样的,均存在其特征曲线。
缓冲溶液的配制:取15. 0 g Na2HPO4 ·12H2O 和1. 59 g KH2PO4 ,加500 mL 蒸馏水配制成pH 7.5 的缓冲溶液备用。果蔬中的农药残留用此溶液提取。
2. 2 实验装置和分析条件
6890 型所相气谱仪(美国安捷伦公司)。图1 是SPME / SnO2 联用技术对农药残留的检测装置图。空气为载气,固定流速为10 mL /s;样品经SPME 进样器解吸进入测试室( 体积为2500 mL);采用HP 6035A 型直流稳压电源和HP 3325B 型信号发生器,输出频率为0.02 Hz 、幅度为5V 的方波信号以保证传感器在一定的温度调制范围工作;信号采集与数据储存由启天2000 6C /1G 微型计算机控制,采集速度2 点/s,2 min 左右完成特征峰测量。
2. 2. 1 萃取条件萃取头类型
PDMS,100 μm;萃取方式:直接浸泡10 min,超声波振荡;解吸时间8 min,温度85℃ ;试样量:15 mL;溶液:4% NaCl(pH 7.5)。
2. 2. 2 色谱条件色谱柱
DB17(30 m ×0.25mm,0.25 μm);载气:He(150 kPa);柱温:50℃(1 min) 20℃ /min升至120℃,再以 5℃ /min 升至280℃ ;进样方式:冷柱头进样;检测器的温度:300℃ ;检测器:FID;OCI 温度:50℃以150℃ /min 升至280℃;压力程序:150 kPa以5 kPa /min 升至350 kPa。
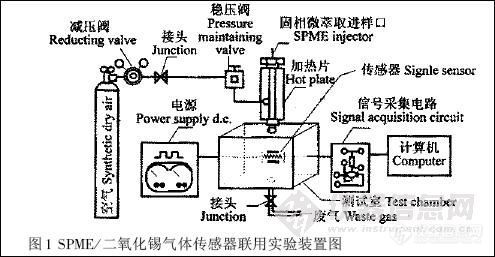
图1 SPME / 二氧化锡气体传感器联用实验装置图
3 结果与讨论
图2 是按照SPME 的操作条件对果蔬中敌百虫、乙酰甲胺磷、乐果、氧乐果、甲胺磷和马拉硫磷进行萃取后,SnO2 气体传感器的动态响应信号。可以看到,各种农药残留在传感器上的动态响应均有其特征峰,2 min 则可以完成4 个周期特征峰的测量。显然,检测速度显著提高。同时,对不同浓度的农药残留,动态响应给出的特征峰的面积也不同。根据不同峰面积可以完成定量分析;也可以根据对特征峰进行FFT,结合极坐标构建完成定量分析[4]。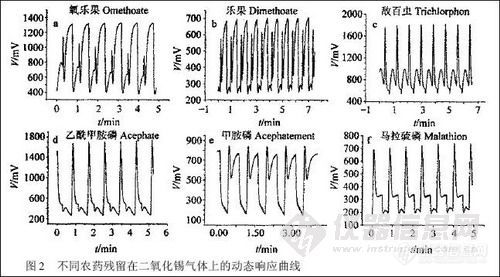
图2 不同农药残留在二氧化锡气体上的动态响应曲线
同样萃取条件下的SPME / gc 分析结果表明,萃取头280℃ 下解吸8 min,6 种农药残留的相对保留时间分别为:甲胺磷7. 11 min,乙酰甲胺磷8. 07 min,敌百虫11. 14 min,氧乐果14. 21 min,乐果16. 55 min,马拉硫磷21. 24 min。显然,用SPME / SnO2 气体传感器联用技术能实现对农药残留的快速检测。
为了分析农药残留在萃取头上的解吸平衡情况,研究了零解吸时间开始测量时传感器对甲胺磷的动态响应信号(如图3 所示,解吸温度为85℃ )。从图中可以看出,大约10 min 左右,在本实验条件下,第3 期黄行九等:固相微萃取/ 二氧化锡气体传感器联用技术对果蔬中有机磷农药残留的快速检测。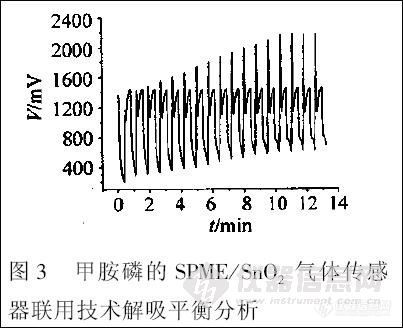
图3 甲胺磷的SPME / SnO2 气体传感器联用技术解吸平衡分析
解吸基本达到平衡。当改变解吸温度,传感器同样可以给出类似的响应信号。同时,在图3 中也可以观察到萃取头的解吸全过程,可以一步得到解吸平衡时间,对研究SPME / SnO2
联用技术的工作条件优化非常有利。而在SPME / gc 、gc / MS 和HPLC 联用技术中,解吸平衡的分析需几步才能完成[9]。
诚然,SPME / SnO2 联用技术对农药残留的快速检测还有许多工作条件,如农药的选择、萃取头的选择、溶液pH 值、萃取温度及萃取时间等有待深入研究。
致谢中国科学技术大学理化实验中心色谱室提供的色谱分析
摘自2005 年3 月Chinese Journal of Analytical Chemistry 363 ~ 365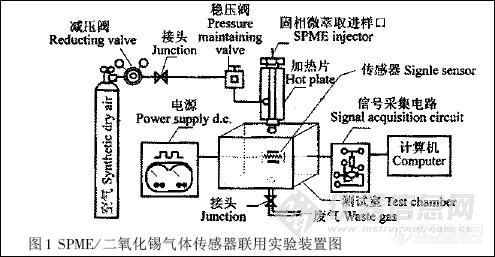
-
+关注
私聊
-
yhl-87_
第44楼2009/11/06
农药残留检验方法系列讲座(39)总结SPE在农残分析中的应用
固相萃取技术在农药残留分析中的应用
摘 要:简介了固相萃取技术的分离模式与作用机理、吸附剂以及操作程序与装置,结合实例对固相萃取技术在农药残留分析中的应用和进展情况进行了评述.
本文由河北大学化学与环境科学学院的刘凡岩教授和马育松研究人员共同完成的河北省科学技术研究与发展计划项目(03547023D); 和保定市科技局攻关项目(03N006)的部分内容。本文综合了固相萃取技术在农药残留分析方面的应用,现全文介绍如下
,
前 言
由于被测残留农药所在基质复杂多样,而且受农药的同系物、异构体、降解产物或代谢产物的影响,样品的前处理成为一个难点和热点. 国内外的分析工作者开发了多种农药残留分析的样品前处理技术[1 ] . 其中固相萃取技术是迅速发展起来的较为实用的前处理技术之一.
固相萃取(solid-phase extraction ,SPE) 技术在20 世纪70 年代出现后,由于其具有操作简便快速、有机溶剂用量少等优点,在农药残留分析的样品前处理中得到了广泛的应用. 本文就固相萃取技术在农药残留分析中的应用做一介绍
.
1 固相萃取技术简介
固相萃取是利用固体附吸剂将样品中的目标化合物吸附,与样品的基体和干扰化合物分离,然后再用洗脱液洗脱或加热解吸附,达到分离和富集目标化合物的目的.固相萃取具有以下特点:固相萃取处理过程中不使用大量互不相溶的溶剂,不产生乳化现象;它采用高效、高选择性的吸附剂,能显著减少溶剂用量;简化了样品处理过程,由于试剂用量少,减轻了环境污染.
1. 1 固相萃取的分离模式及作用机理
固相萃取的主要分离模式可分为正相、反相、离子交换和吸附. 其作用机理包括氢键、偶极作用、疏水性相互作用和静电吸引力等[2 ] .
1. 2 固相萃取所用吸附剂的选择
由于农药品种众多,性质差异较大,因此用于农药残留分析的固相萃取吸附剂种类也较多,吸附剂种类的选择依样品的性质而定.
1. 2. 1 若样品的基质是水溶液
a) 分析目的物更易溶于水相,若带电荷则选择离子交换柱,如阴离子或氨基柱(适于阴离子或酸性化合物) ,阳离子交换柱(适于阳离子或碱性化合物) ;若为中性则选择反相柱或强阴(阳) 离子柱.
b) 分析目的物更易溶于有机相,若带电荷则选择离子交换柱或反相柱;若为中性则选择反相柱.
1. 2. 2 若样品基质是有机相
a) 若是极性有机相,用水稀释,按水溶液方案进行.
b) 若是中等极性到非极性有机相,选择正相柱或将溶剂挥发至干后,再溶于水或极性溶剂,按水溶液方
案进行.
1. 3 固相萃取的操作程序及装置
1. 3. 1 固相萃取的操作程序
尽管固相萃取的分离模式多种多样,但其操作程序基本类似,一般由下面3步组成.
a) 条件化:在萃取样品前用适当的溶剂淋洗固相萃取小柱,使吸附剂保持润湿,可以吸附目标化合物或干扰化合物.
b) 加样:将液态的样品倒入已条件化的固相萃取小柱,视样品情况不同,可利用重力、抽真空、加压和离心等不同形式使样品进入吸附剂.
c) 洗涤和洗脱:视分离模式和样品性质,可采用适当的洗脱剂将目标化合物直接淋洗下来;也可先将干扰化合物淋洗掉,再用适当的洗脱剂将目标化合物洗脱. 通常采用后一种方法更有利于样品的净化

图1 真空多歧管固相萃取装置
1. 3. 2 固相萃取的装置
常用的固相萃取装置有固相萃取管和片. 固相萃取管是一根由聚丙烯、聚乙烯、聚四氟乙烯或不锈钢制成的小柱,下端装有筛板以支撑吸附剂,在吸附剂上再加一筛板以防止加样时破坏柱床;固相萃取片则是将吸附剂埋于玻璃棉中,制成圆盘状.
目前,多种规格的装有不同种类吸附剂的商品小柱已普遍面市,相关厂商为配合固相萃取柱的使用,开发了固相萃取的专用装置:有给单个固相萃取柱加压的单管处理塞,也有可同时进行多个固相萃取柱抽真空操作的真空多歧管装置(图1) . 在实际应用中,分析工作者还开发了可进行自动固相萃取的实用装置,如Colume 等利用连续固相萃取装置(图2) 测定了蔬菜中的有机氯和拟除虫菊酯类农药残留[3 ] ,此装置的使用,不仅简化了操作步骤,减少了样品前处理的时间,降低了溶剂的消耗,而且固相萃取柱经清洗后可多次继续使用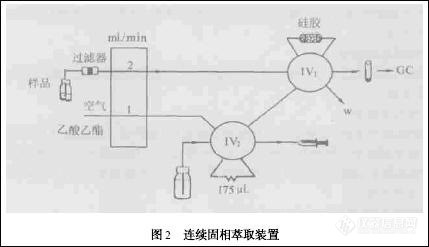
2 固相萃取技术在农药残留分析中的应用
国内外分析工作者就固相萃取技术在农药残留分析中的应用方面进行了广泛的尝试和探索,取得了许多成功的经验. 由于农药在农作物生产中不仅污染作物本身,对农作物的生长环境也产生污染,包括土壤、水体等. 下面结合实例对固相萃取技术在水、土壤、食品中的农药残留分析中的应用进行简介.
2. 1 SPE 在水体中农药残留分析方面的应用
测定水体中的农药残留一般采用如C18 ,C8 等非极性吸附剂,通常用甲醇和水条件化,以甲醇为洗脱剂.由于对水体中的农药残留限量要求严格,如欧盟规定地表水农药残留量为1. 0μg/ L ,饮用水为0. 1μg/ L[4 ] ,我国规定生活饮用水中滴滴涕、六六六的限量分别为1μg/ L 和5μg/ L[5 ] ,而且自然水体中的农药残留质量浓度通常也很低,若没有可靠的分离富集手段很难检测到,采用固相萃取技术可以使提取、富集和净化一步完成. 将大体积样品过固相萃取柱进行预浓缩,用小体积洗脱剂洗脱,再浓缩定容进行检测,大大降低了检测方法的检出限. 如:康跃慧等人测定水源水中有机磷,通过固相萃取富集分离后,使方法检出限达到了1. 19~
5. 34 ng/ L[6 ] ;Lopez-Blanco 等测定地表水中的硫丹(α和β异构体) ,采用固相萃取实现了样品的100 倍浓缩富集,使方法检出限达到20 ng/ L[7 ] ;Pinto 等测定水中草净津等4 种除草剂,采用固相萃取富集样品使浓缩倍数达到500 倍,方法检出限降低至9. 8~34 ng/ L[8 ] .
但对于水溶性强的农药品种如甲胺磷、乐果、敌敌畏等,回收率仅50 %~60 % ,甚至更低[6 ,9 ] ,因此在今后的工作中应着眼于如何提高这些农药品种的回收率,提高方法的准确度
2. 2 SPE 在土壤中农药残留分析方面的应用
测定土壤中的农药残留一般是先用适当的提取溶剂及提取方式从土壤样品中将待测的农药提取出来,再利用固相萃取技术进行净化. 由于待测农药的性质不同所使用的提取溶剂不同,因此从基质中带来的杂质性质也不尽相同,所以要选择适当的吸附剂实现待测残留农药的分离和净化.
分析极性较强的农药,采用极性较强的提取溶剂如丙酮-水体系时可采用非极性吸附剂如C18等,这样提取溶剂中水溶性强的杂质不会保留在吸附剂上,有利于样品的净化,如Ruiz 等利用V (水) ∶V (DMF) = 100∶2.5 为提取溶剂、C18为吸附剂,以乙酸乙酯为淋洗剂测定土壤中莠去津,呋喃丹,地亚农等农药取得了较好的效果[10 ] ;而分析极性较弱的农药,采用极性较弱的提取溶剂时,可以选择极性吸附剂如Florisil 等,它不会对提取溶剂中的弱极性杂质产生保留,然后再选择适当溶剂将待测残留农药淋洗下来进行测定,如Kim 等利用V(正己烷) ∶V (二氧甲烷) = 7∶3 为提取溶剂、Florisil 为吸附剂测定土壤中α,β-HCH ,七氯等有机氯农药[11 ] .
2. 3 SPE 在食品中农药残留分析方面的应用
食品种类很多,基质类型复杂多样,干扰物质也不尽相同,其中包含各种色素、不同比例的脂肪和水分等. 因此针对不同类型样品、不同的待测农药选择不同类型的吸附剂. 对于液态样品可直接或经简单稀释后,用固相萃取柱进行提取和净化[12 ] ;而固体样品可先用适当方式提取后,再用固相萃取技术净化
.对于多水分低脂肪的蔬菜和水果样品,一般先用极性溶剂如乙腈、丙酮等溶剂提取,然后根据所分析农药的种类和性质,选择相应固相吸附材料净化分离;对于低水分低脂肪的粮食类样品,则先加入一定量的蒸馏水润湿后,再用溶剂提取,过固相萃取柱净化,然后测定;对于油脂类样品,加入与样品等量的丙酮,混匀,用己烷和乙腈进行液-液萃取,或加入乙腈后,零下20 ℃冷冻去脂,再进行固相萃取[18 ] . 具体应用实例见表1.
-
+关注
私聊
-
yhl-87_
第45楼2009/11/06
农药残留检验方法系列讲座(39)(下)总结SPE在农残分析中的应用
3 固相萃取新应用
3. 1 多种固相萃取吸附剂的联用
在农药残留分析的样品前处理中所面对的基质和干扰化合物复杂多样,性质差异很大,采用单一种类的SPE 吸附剂有时难以有效去除基质和干扰化合物,因此分析工作者将2 种SPE 吸附剂联用进行样品前处理.戴华等采用乙腈提取,ENVI-CARB 和LC-Alumina-N 固相萃取小柱串联净化,HPLC-DAD 测定了稻谷中的吡虫啉农药残留量[19 ] ;李拥军等用V (丙酮) ∶V (正己烷) = 1∶1 提取茶叶中的甲氰菊酯、氯菊酯等5 种拟除虫菊酯类农药,采用活性碳和中性氧化铝柱串联使用,有效去除了茶叶中的色素、有机碱和酚类化合物等干扰成分[20 ] ;林维宣等利用HPLC-UV 同时检测大米中的杀线威、灭多威等8 种氨基甲酸酯类农药残留量时,采用丙酮提取,Florisil 和C18双柱净化样品,提高了检测灵敏度[21 ] ;Rastrelli 等利用GC-NPD 测定橄榄油中的甲胺磷、地亚农、对硫磷等18 种有机磷农药残留量,采用硅胶柱和C18柱串联的方法实现提取和净化一步完成[22 ] .
3. 2 固相微萃取
固相微萃取(Solid phase micro-extraction ,SPME) 是20 世纪80 年代末由加拿大Waterloo 大学Pawliszyn 和Arhturhe 教授提出的,它是在固相萃取技术上发展起来的一种兼样品制备的前处理技术. SPME 的原理是利用待测物在基体和萃取相之间的非均相平衡,使待测组分扩散吸附到石英纤维表面的固定相涂层,待吸附平衡后,再以气相色谱或高效液相色谱分离和测定待测组分. SPME 技术具有简便、快捷、无需萃取溶剂的特点,非常适用于农药残留分析. 影响固相微萃取结果的因素包括萃取头类型、萃取时间、离子强度、解吸附温度和解吸附时间等[23 - 24 ] . 陈伟琪等利用100μm 聚二甲基硅氧烷萃取头浸入匀浆液的萃取方式,GC- FPD 测定了茼蒿中的甲拌磷等4 种有机磷农药残留量,讨论了搅拌速度、离子强度、平衡时间等影响因素[25 ] ;Lam2bropoulou 等采用100μm 聚二甲基硅氧烷萃取头,顶空-固相微萃取(HS-SPME) 方式,GC-MS 方法测定了草莓和樱桃中的乙硫磷、对硫磷、倍硫磷等7 种有机磷农药残留量[26 ] ;Sakamoto 等发展了一种采用85μm聚丙烯酸酯萃取头、顶空-固相微萃取测定水中的158 种农药残留量的GC-MS 方法,探讨了不同种类萃取头、盐的浓度、pH 值和萃取温度等参数的影响[27 ] .
3. 3 基质固相分散萃取
基质固相分散萃取(Matrix solid-phase dispersion extraction ,MSPD) 是由Barker 于1989 年首次提出,这项技术的优点是不需要进行组织匀浆、沉淀、离心、pH 调节和样品转移等操作的步骤.MSPD 的原理是将C18等固相萃取吸附剂与固体、半固体或黏性的样品一起研磨,得到半干状态的混合物并将其作为填料装柱,然后用不同的溶液淋洗柱子,将各种待测物洗脱下来.MSPD 多应用于从动物组织、蔬菜、水果中分离药品、除草剂、杀虫剂和其他污染物,影响MSPD 效果的因素包括吸附剂的粒径、键合相的性质、样品基质的性质以及基质
的修饰、淋洗剂的性质以及加入的顺序等[28 - 29 ] . Kristenson 等发展了一种简便快速的MSPD/ GC-MS 方法测定了水果中的氯菊酯和倍硫磷、马拉硫磷等10 种有机磷农药残留量,选择C8 作为吸附剂,每次分析仅需25 mg样品和100μL 溶剂[30 ] ;Pico 等利用C18做分散吸附剂,硅胶柱净化,GC-NPD 和GC- ECD 测定的方法检测了水果和蔬菜中的克菌丹、灭菌丹等8 种杀菌剂[31 ] ;Morzycka 建立了一种简便的多残留分析的MSPD/ GC-NPD 方法测定蜂蜜中的乙硫磷、毒死蜱等12 种农药的残留量,采用Florisil 和硅胶作分散吸附剂,氧化铝和硅胶作为净化吸附剂,达到了较好的净化效果和良好的回收率[32 ] .
3. 4 新型固相萃取吸附剂
随着新型农药品种的不断涌现,固相萃取所用的吸附剂也得到了相应的发展,相继出现了一些新型吸附剂.Queiroz 等发展了一种“绿色化学”方法,采用γ射线辐照的方法将聚甲基十八烷基硅氧烷固定在硅胶上作为固相萃取的吸附剂,合成过程中不产生有毒废物,用该吸附剂萃取水中的农药残留取得良好的回收率[33 ] ;Rejeb 等将抗体固定在硅胶吸附剂上制成免疫亲和柱用于萃取花生中的满穗,取得良好的选择性和满意的回收率[34 ] .
4 结语
固相萃取技术问世以来得到了长足的发展,并在农药残留分析中得到广泛的应用,随着新型吸附剂和新的操作模式的不断涌现,固相萃取技术在农药残留分析中的应用将更具生命力.


-
+关注
私聊
-
yhl-87_
第47楼2009/11/06
农药残留检验方法系列讲座(40)在动物组织中用HPLC-MS/MS分析16种兽药
高效液相色谱的-串联质谱法测定动物组织中
16种喹诺酮类药物残留
摘 要:建立了动物组织样品中萘啶酸、恶喹酸、氟甲喹、诺氟沙星、依诺沙星、环丙沙星、洛美沙星、丹诺沙星、恩诺沙星、氧氟沙星、沙拉沙星、二氟沙星、麻保沙星、培氟沙星、司帕沙星、奥比沙星等16种喹诺酮类兽药多残留量的高效液相色谱一串联质谱测定方法。用酸性乙睛萃取样品中的16种喹诺酮类药物残留,然后用正己烷脱脂,旋转蒸发浓缩,以Inertsil C8-3色谱柱分离,在正离子模式下以电喷雾电离串联质谱进行测定。在10,50,100 μg/kg 3个加标水平下进行了验证试验,方法的线性范围为10~100μg/kg,平均回收率为62.4%~102 %,相对标准偏差为1.4%~11.9%。该方法简便、快速、准确,各项技术指标满足国内外法规的要求,可用于鸡肉、鸡肝和鱼肉等动物组织样品中喹诺酮类药物多残留的确证检测
本文由岳振峰1, 林秀云1, 唐少冰1, 陈小霞2, 吉彩霓1, 华红慧1, 刘昱1
(1.深圳出人境检验检疫局食品检验检疫技术中心, 2.深圳大学教务处) 等研究人员共同完成的兽药喹诺酮类残留的HPLC-MS/MS方法的分析
前 言
喹诺酮类 抗菌药(quinolones,QNs)是指人工合成的含有4一喹酮母核的一类抗菌药物,其中氟喹诺酮类药物(fluoroquinolones)已逐渐成为该类药物的主流 [1]。因其具有抗菌谱广、高效、低毒、组织穿透力强、价格低廉等特点,目前喹诺酮类药物已成为兽医临床诊断和水产养殖中最重要的抗感染药物之一,被大量用于治疗和预防动物疾病及促生长,国内兽用喹诺酮类药物的使用量已接近500吨/年。在食品动物饲养中较常用的喹诺酮类药物包括萘啶酸、恶喹酸、氟甲唆、诺氟沙星、环丙沙星、丹诺沙星、恩诺沙星、氧氟沙星、沙拉沙星、二氟沙星、麻保沙星、奥比沙星等。喹诺酮类药物同时也是人类最有效的抗菌药之一,由于细菌对喹诺酮类药物耐药性的产生和某些喹诺酮类药物的潜在致癌作用,其残留问题已引起人们的广泛关注。美国禁止在食品动物养殖中使用氟喹诺酮类药物;我国及世界卫生组织、欧盟、日本等国家和组织都将喹诺酮类药物列入限制使用的兽药名单中,并制订出相应的最高残留限量:根据不同动物品种、组织和药物种类,最高残留限量在10~6000μg/kg之间。日本2006年5月29日开始实施“肯定列表制度”后,对喹诺酮类药物残留的检测要求进一步提高。
关于喹诺酮类药物残留的检测方法,目前多采用杯碟法[1]、高效液相色谱一荧光法(HPLC-FLD)[2~7]、HPLC-串联质谱(Ms/Ms)[8~15]等。由于HPLC-MS具有选择性强、灵敏度高和一次可检测多种喹诺酮类药物残留的优点,在喹诺酮类药物的确证分析中应用较多。本文在国内外研究的基础上,利用HPLC一MS/MS技术,建立了动物组织样品中16种喹诺酮类药物残留的同时确证分析方法。
实 验
1 实验部分
1.1 仪器与材料
Agilent 1100型高效液相色谱仪,API3000型电喷雾(ESI)离子源串联质谱仪;Minishaker MSI型[漩涡混合器; Universal 32R型低温离心机; Laborota4003型自动旋转蒸发仪。萘啶酸(纯度9.5%)、诺氟沙星(纯度98%)、依诺沙星(纯度98%)、洛美沙星(纯度99.5%)、氧氟沙星(纯度99%)购自Sigma公司; 恶喹酸(纯度97.0%)、环丙沙星(纯度98%)、恩诺沙星(纯度98.0%)购自Fluka公司; 氟甲喹(纯度99.7%)、麻保沙星(纯度9.8%)、奥比沙星(纯度99.8%)购自Riedel一de Haen公司; 丹诺沙星(纯度100%)购自Sequoia Research Products Ltd.; 沙拉沙星(纯度98.0%)、二氟沙星(纯度99%)、培氟沙星(纯度99.0%士0.5%)、司帕沙星(纯度99.5%士0.5%)购自Dr..E公司。甲醇、乙睛、甲酸为色谱纯试剂; 其他试剂均为分析纯试剂; 水为高纯水。鸡肉、鸡肝、鱼肉为市购。
1.2 HPLC一MS/MS条件
1.2.1 色谱条件
色谱柱 : Inertsil C8一3柱(5μm, 15Omm×2.1mm);柱温:30℃;进样量:30μL。流动相A:0.1%甲酸乙睛溶液;流动相B:0.1%甲酸水溶液; 洗脱梯度:流动相A的比例在6min内由13%线性提高到90%,然后保持4min;柱平衡时间8min,流速:0.3mL/min。
1.2.2 质谱条件
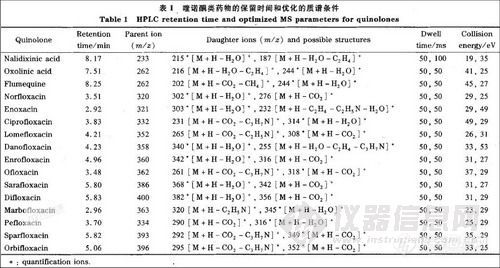
离子化模式: ESI(十);质谱分辨率:单位分辨率;雾化气流速:氮气6.00L/min; 气帘气流速:氮气12.00L/min;喷雾电压:500V;去溶剂温度: 500℃;去溶剂气流速:氮气7.00L/min;碰撞气流速:氮气6.00mL/min。其他质谱分析参数见表1。
1.2.3 样品处理
称取均质试样5g (精确到0.01g),置于50mL聚丙烯离心管中,用15mL 1%乙酸乙睛溶液于15℃以下超声提取5min,于15℃、3500r/min下离心5min,收集上层清液并置于另一干净的离心管中。重复上述提取步骤1次,合并上层清液,加人15mL正己烷,混合lmin,于15℃、3500r/min下离心5min,弃去正己烷层。将下层 (乙睛层) 转移至100mL鸡心瓶中,加人5mL异丙醇,混合均匀,于45℃下减压蒸发至近干。用流动相(13%流动相A和87%流动相B的混合液)将残渣分次转移至15mL具刻度离心管中,定容至5mL。加入5mL正己烷混合lmin,于5℃、3500r/min下离心5min。弃去上层(正己烷层),将下层用0.45μm
滤膜过滤,待测定。
结果与讨论
2.1 色谱分离条件的优化
2.1.1 色谱柱的选择
因喹诺酮类化合物的叔胺基和竣基官能团能在水中发生解离,色谱柱固定相表面的残存硅醇基和金属离子可通过氢键或离子交换作用强烈吸附喹诺酮类化合物,出现色谱峰拖尾、保留时间不稳定或过长,甚至被保留在色谱柱上,导致峰形异常和分离度下降。因此需要选用以高纯硅胶为基质并经端基封闭处理的C18柱、C8柱、苯基柱或非硅胶基质的聚合物反相固定相作为分离柱。本文采用Inertsil C8一3柱进行了分离试验,发现该柱对喹诺酮类化合物具有良好的分离效果和对称的峰形,可满足质谱检测的要求。
-
+关注
私聊
-
yhl-87_
第48楼2009/11/06
农药残留检验方法系列讲座(40)(下)在动物组织中用HPLC-MS/MS分析16种兽药
2.1.2 流动相的选择与优化
流动相的pH值对喹诺酮类化合物的分离和保留具有显著影响:
(1) 喹诺酮类化合物为酸碱两性化合物,其解离状态和在流动相中的溶解性随流动相的pH值而变化;
(2)硅胶基质键合固定相表面的残余硅醇基的解离程度与流动相的pH值有关,pH>3即可完全解离。为维持流动相的低pH值以抑制硅醇基的解离和保证喹诺酮类化合物在流动相中的稳定溶解状态,本文采用0.1%甲酸或乙酸来控制流动相的pH值,试验发现二者均可得到对称、尖锐的峰形,但甲酸更利于喹诺酮类化合物的离子化,可得到较高的检测灵敏度,故本文选择用0.1%甲酸控制流动相的_pH值。流动相 中有机改性剂的含量也对喹诺酮类药物的分离有重要影响:有机改性剂的含量过高,喹诺酮类化合物在色谱柱上的保留过弱,流出过快,达不到分离度要求甚至发生簇集现象;有机改性剂的含量过低,则喹诺酮类化合物在色谱柱上的保留过强,特别是极性较弱的萘啶酸、恶喹酸和氟甲喹的保留时间太长,易发生色谱峰拖尾现象。本文经反复试验,发现以0.1%甲酸乙睛溶液和0.1%甲酸水溶液为流动相,采用梯度洗脱方式可实现喹诺酮类化合物的快速分离。
2.2 质谱测定条件的优化
首先采用 l mg/L的喹诺酮类化合物的标准溶液以流动注射的方式在正离子模式下进行母离子全扫描,确定各种喹诺酮类化合物的分子离子,然后分别以其分子离子为母离子,对其子离子进行全扫描。喹诺酮化合物的二级质谱中主要的碎片离子峰是喹诺酮药物分子的脱水峰([M+H一H20]+)、脱羧峰(〔M+H一CO2〕+)以及脱梭后哌嗪环断裂发生结构重排失去C2H4NR的产物离子([M+H-CO2-C2H4NR]+) (见表1)。最后以多反应监测(MRM)正离子模式优化了各种质谱分析参数。优化得到的最佳质谱条件如“l.2.2”节及表1所述。
2.3 提取与净化条件的优化
喹诺酮类化合物的分子结构中含有羧基和叔胺基,具有酸碱两性性质,易溶于酸性或碱性溶液。文献[1] 报道的提取方法包括用纯有机溶剂 (乙睛、甲醇、二氯甲烷等),或有机溶剂添加一定比例的酸或碱等提取,含水的乙睛或甲醇可显著提高回收率。本文在国内外研究的基础上,采用不同比例的1%乙酸水溶液和1%乙酸乙睛溶液混合液作为提取溶剂进行提取试验,结果表明,水溶液的比例越高,提取液中的蛋白质含量越高,提取液澄清和浓缩越困难,而完全采用1%乙酸乙睛溶液作为提取溶剂不仅除蛋白效果好,而且容易浓缩,提取回收率也可满足要求。这可能与组织样品本身都含有一定的水分有关。采用浓缩前后两次正己烷脱脂处理可得到澄清透明的样品溶液,能够满足净化要求。
2.4 样品基质效应的消除
大气压喷雾电离离子源质谱分析容易受样品基质的影响。试验中发现,样品基质对离子化有非常强的抑制或促进作用,不同样品基质对各种喹诺酮类抗生素药物离子化的抑制情况存在显著的差异。为消除样品基质效应,以空白样品提取液作为标准溶液的稀释溶液,可使标准和样品溶液具有同样的离子化条件,从而消除了样品基质效应。
2.5 方法的线性范围与测定低限
用空白样品提取液逐级稀释标准混合工作液进行HPLC一MS/MS分析,测定结果用Microsoft Excel软件进行回归处理,16 种喹诺酮类药物的线性范围均为10一100μg/kg,测定低限均为10μg/kg。
2.6 方法的回收率与精密度
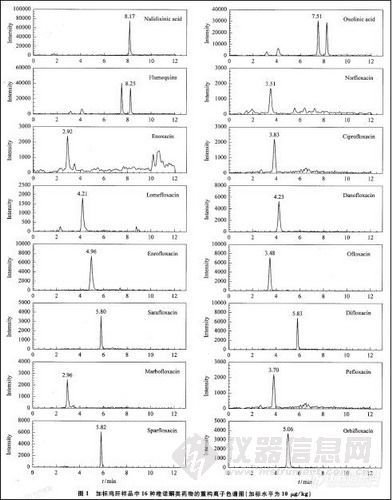
选用不含 16种待测组分的鸡肉、鸡肝和鱼肉样品进行100,50,10μg/kg 3个添加水平的回收试验,每个添加水平平行测定10个样品,方法的平均回收率:鸡肉71.9%一102%,鸡肝6.4%一102%,鱼肉62.4%一89.6%;相对标准偏差:鸡肉1.5% -11.9%,鸡肝2.7%一9.1%,鱼肉1.4%一11.4%。这些指标满足国内外对残留分析的要求。加标鸡肝样品的16种喹诺酮类药物的重构离子色谱图见图1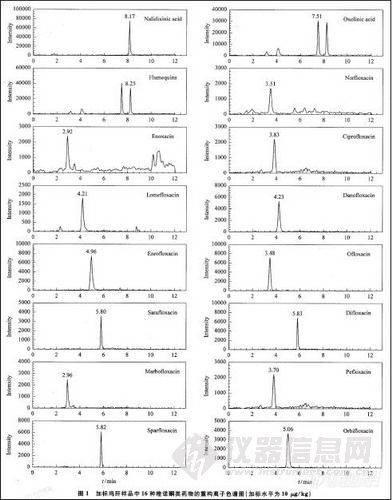
3 结论
本文采用酸性乙睛溶液提取、正己烷脱脂、高效液相色谱分离、串联四极杆质谱检测,成功建立了动物组织样品中16 种喹诺酮类药物残留的高效液相色谱一串联质谱测定方法。方法简便、快速、准确,方法的测定低限为10μg/kg,达到目前国际上检测喹诺酮类药物残留的先进水平,可用于鸡肉、鸡肝和鱼肉等动物组织样品中16 种喹诺酮类药物残留的确证检测。

-
+关注
私聊
-
yhl-87_
第49楼2009/11/06
农药残留检验方法系列讲座(41)GC-μECD分析茶叶中巴丹残留
气相色谱-微池电子捕获检测器分析茶叶中杀螟丹的残留
摘 要:介绍了一种快速、灵敏、可靠的适用于茶叶样品中杀螟丹残留量的气相色谱分析方法、茶叶样品经过0.05mol/L盐酸溶液提取,液-液分配净化,在碱性条件下用正己烷萃取提取液中的杀螟丹,用毛细管气相色谱法-微池电子捕获检测器(u ECD)测定杀螟丹的残留量。结果表明:样品的添加回收率为70.2%~92.0%,相对标准偏差小于l0%,方法的检测低限为0.01 mg/kg,符合农药残留分析的要求。
本文是浙江省重点科研社会发展资助项目,由吴 刚1,2, 虞慧芳3, 鲍晓霞1, 陈 浩1, 叶庆富2(1浙江出人境检验检疫局,浙江省食品安全重点实验室;2浙江大学原子核农业科学研究所;3浙江省农业科学院蔬菜研究所)共同完成。
前 言
杀螟丹(cartap),中文商品名为巴丹,化学名为1,3-二(氨基甲酰硫)-2-二甲基氨基丙烷。杀螟丹是沙蚕毒素类杀虫剂,具有内吸胃毒、触杀等多种作用,效果迅速,持效期较长,杀虫谱广,具有较长的残效期[1],使用较广泛[2]
欧盟新规定茶叶中杀螟丹的最大残留限量(MRL)值为0.1 mg/kg,日本等国也对茶叶中的杀螟丹残留提出了检测要求[3]。由于该化合物难以气化,不能直接用气相色谱法(GC)测定,目前国内外关于杀螟丹残留分析方法的报道仅限于样品基质比较简单的土壤、蔬菜等样品,而对于样品基质较为复杂的茶叶中杀螟丹的残留分析方法较少[4~8]。笔者[9]曾利用衍生反应将杀螟丹转化为沙蚕毒素,采用气相色谱-火焰光度检测器(GC-FPD)测定,但是该方法实验过程较为繁琐,样品处理周期较长。
杀螟丹盐酸盐在碱性条件下转化成分子状态杀螟丹,用正己烷提取后,可以用气相色谱进行测定。基于此,本研究提出了一种适用于茶叶样品中杀螟丹残留检测的新方法,即用正己烷对茶叶的水解产物进行萃取,调节溶液的pH值,在碱性条件下进行液一液分配净化,最后采用毛细管气相色谱法(配微池电子捕获检测器(μ-BCD))进行分析。该方法能有效地减少样品中杂质的干扰,操作十分简便、快捷,灵敏度高,回收率和检测低限均能满足国内外的检测要求。
1 实验部分
1.1 仪器与试剂
Agilent 6890 N气相色谱仪(带μ-EOD,Ni63),DB-1701毛细管色谱柱(30 m×0 25 mm×0.25μm),超声波仪,旋转蒸发仪(Bruchi R205),离心机,微型高速试样粉碎机(rvc80,天津市泰斯特仪器有限公司),pH计。
杀螟丹盐酸标样(纯度99.9%、活性炭、正巳烷、HCI、NaCl、NaOH、NaHCO3、无水 Na2SO4(650℃烘4 h.保存在干燥器中)等均为分析纯茶叶样品(皇帝牌龙井茶,浙江嵊州,浙江华发出口茶厂)
1.2 分析方法
1.2.1 提取和净化
准确称取2.0 g(精确到±0.0l g〉茶叶粉末于50 mL具寒离心管中,加人40 mL 0.05 mol/L盐酸溶液,在80℃水浴中超声提取20 nim,然后在4 000r/min离心5 min:将上清液转入另一50ml具塞离心管中,用0.05 mol/L盐酸溶液定容为40 ml,取20rnL加入15 mL 正己烷和0.1 g活性炭。涡旋提取1 min,于4 000 r/min离心5 min;将上层有机相{用吸管吸弃.再加入15 mL 正已烷,重复一次。将萃取后的溶液过滤到另一具塞离心管中,然后用2mol/L的NaOH调节pH值至8.5~9.0,加入5 mL50 g/L的NaHCO3溶液.再加入4 mL正已烷.涡旋提取1 min,于4 000r/min离心5 min;将上层有机相用吸管转移到进样瓶中,供气相色谱测定。
1.2.2 气相色谱检测条件
u-ECD温度:280℃;进样口温度:250℃;卡}:注温程序升温条件:初始值为70℃,保持1 min,然后以l0℃/min的速率升至20.0℃,保持l0 min,Postrun 270℃(5 min);载气:高纯氮(99.999 9%),流速1 ml/min;尾吹气:30 ml/min:进样量:1.0 μL
2 结果
2,1 方法线性关系的确定
用去离子水配制0.1,0.3,0.5,0.7,I.0 mg/L的系列杀螟丹标准溶液,经反应、萃取和'气相色谱法测定.杀螟丹进样浓度的对数值(x)与峰面积响应值的对数值(y)之间呈线性相关,线性方程为y=2 155.9x+40.l,相关系数(r2)为0.997 2,表明相关性较好,
2.2 方法的检测灵敏度
在上述样品处理方法及色谱条件下,本方法杀螟丹的最低检出量为0.007 ng,测定低限为0.01mg/kg,能够满足检测灵敏度的要求。杀螟丹的标准图谱及空白样品与添加样品的色谱图见图1~3。可以看出,杀螟丹标样的峰形较好,响应值也较高,优于文献[9]中杀螟丹的峰形和响应值[9]。虽然茶叶样品中含有很多杂质,但由于采用活性炭去除了部分色素等杂质,并通过调节溶液的酸碱度,在色谱分析中运用程序升温技术,可使杂质峰与样品峰明显分离。
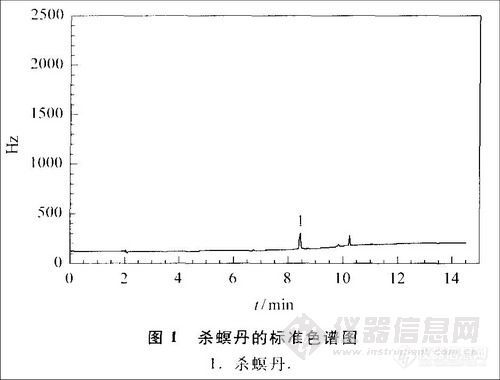
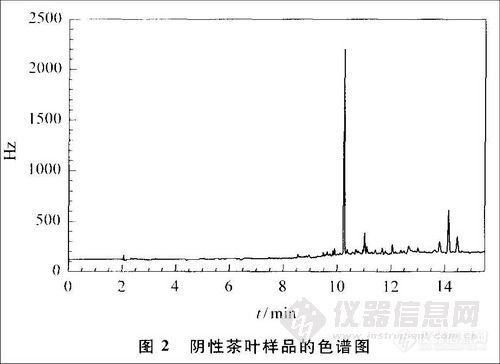
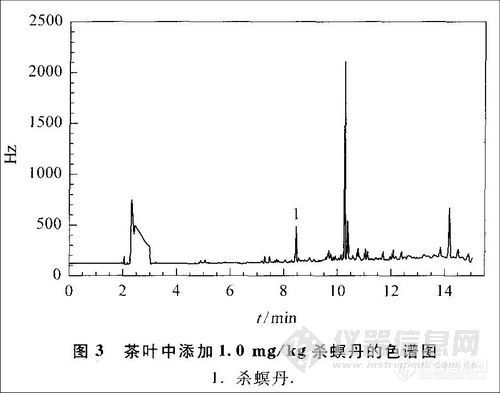
2.3 添加回收率试验与精密度试验
称取2.0 g茶叶阴性样品,分别添加0.1,0.5和1.0 mg/kg的杀螟丹标准溶液(每个浓度制备6个样品),按照上述的提取、净化与色谱条件进行实验,测定茶叶样品中的杀螟丹含量,计算回收率及其相对标准偏差(RSD)(见表1),结果表明该法满足残留分析要求。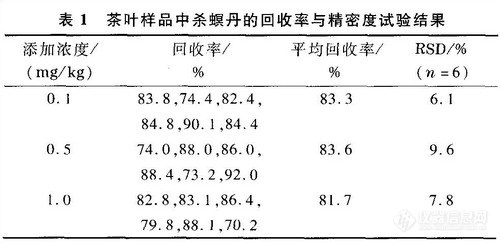
3 讨论
杀螟丹属内吸性的农药,在酸性条件下较稳定,以盐酸盐的离子状态存在,pH值超过9时极易分解。茶叶样品的基质成分较为复杂,为了最大限度地提取出茶叶中的杀螟丹,而尽可能少地引人杂质,采用0.05 mol/L盐酸溶液作为提取液,在80℃水浴中超声提取,大大提高了提取效率。在样品净化过程中,加入活性炭到提取液中,用正己烷萃取2次后,将有机相弃去,然后小心调节溶液pH值至8.5~9.0后,再用正己烷萃取后直接进样。提取产物与大部分杂质能够较好的分离,有效地降低了本底干扰。整个过程非常简便,方法准确、可靠。
-
+关注
私聊
-
yhl-87_
第50楼2009/11/06
农药残留检验方法系列讲座(42)SPE-衍生化GC-FID测鼻咽剌激剂
固相萃取衍生化气相色谱法测定水中二苯氰胂
摘 要:建立了水中二苯氰胂的检测方法。水样经巯基乙酸甲酯衍生、ODS-C18柱富集、甲苯洗脱后用气相色谱/氢火焰检测器(GC-FID)进行定性定量测定[1]。二苯氰胂的方法回收率在95.3%~104.2%之间,相对标准偏差为2.9%,检出限为20pg。
前 言 二苯氰胂(DC)是第一次和第二次世界大战期间大量生产和使用的一种鼻咽刺激剂(刺激阈值为0.005mg/m3)。侵华日军在我国境内遗弃的化学武器中有相当数量的二苯氰胂,由于埋藏地下五十多年,已有部分的腐蚀泄漏,对当地环境和人民生活已造成了不同程度的危害。国际癌症研究机构(IARC)研究认为砷和砷的化合物属Ⅰ组人类致癌物,即使水中有微量的污染也会通过生物富集对人的机体造成伤害。因此,建立一种灵敏的水中二苯氰胂的测定方法十分必要。
本文由防化指挥工程学院化学侦检分析教研室的曲刚莲 、马果花 、岳丽君、 李善茂等老师们,建立了对致癌物鼻烟剌激剂二苯氰胂的分析方法,现全文介绍如下:
实 验
1 实验部分
1.1 仪器与试剂
6890 型气相色谱仪(Agilent),带有氢火焰离子化检测器(FID);配有30m×0.25mm×0.25μm HP-5 弹性毛细柱;色谱工作站处理系统;载气为N2:99.999%,H2:99.99%,Air;SHB-Ⅲ循环水式多用真空泵;超声清洗器;ODS-C18固相萃取小柱(Agilent 公司,AccuBond SPE ODS-C18 Cartridges 500mg,6mL)。DC 染毒水样,中国环境科学研究院提供;巯基乙酸甲酯(TGM),SIGMA公司;甲苯、乙酸乙酯、丙酮均为HPLC 级,TEDIA 公司。
1.2 色谱条件
进样口温度250℃;起始柱温70℃,保持1min,以30℃/min 升温速率升至280℃,保持3min;检测器温度为270℃,尾吹气流速为60mL/min;载气流速为1.5mL/min,不分流进样。
1.3 实验方法?
取一定量的染毒水样20mL,用0.5M 的硫酸调pH 值为4 左右,加入10μL衍生化试剂巯基乙酸甲酯,摇匀,60℃条件下衍生10min,冷却后用ODS 萃取柱(SPE)进行萃取。将ODS 萃取小柱置真空固相萃取装置上,加5mL 甲醇于柱内,真空抽滤,再用5mL 蒸馏水洗涤一次,但不能使吸咐剂抽干。将衍生好的水样倒入萃取小柱,调节萃取装置真空旋钮,以0.5~1mL/min 流速通过小柱,小柱置于离心机内以3000r/min 离心10min,再放到真空固相萃取装置上,准确移取甲苯2mL 进行洗脱,流速为0.5mL/min,取出收集的洗脱液经无水硫酸钠干燥后供GC/FID 分析用。
2 结果与讨论
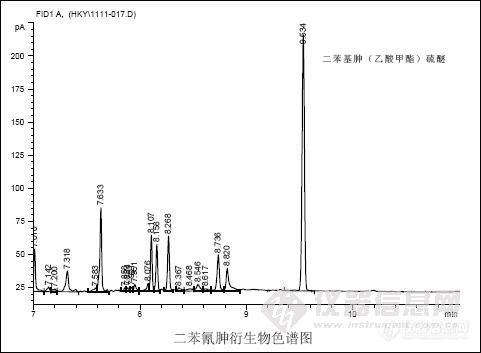
2.1 定性分析
在选定的条件下衍生二苯氰胂水溶液,进样量1.0μL。色谱图如下。在本分析条件下,二苯氰胂衍生物的出峰时间是9.534min。ddd
2.2 洗脱剂的选择及用量的确定
样品溶液是水样,水在反相萃取体系中是弱溶剂,不会影响目标化合物在吸附剂上的吸附,故采用ODS-C18 萃取柱进行反相固相萃取[2]。以甲苯、乙酸乙酯、丙酮作洗脱剂进行筛选。经试验测定,甲苯作洗脱剂时衍生化产物的色谱响应值较高,见表1,因此,选定甲苯作为该实验方法的洗脱剂,洗脱剂的用量为2mL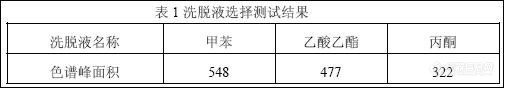
2.3 衍生化条件的选择
2.3.1 温度对衍生化的影响
为使衍生化尽可能的完全,对衍生化温度进行了选择。在20℃、40℃、60℃、80℃四种不同温度条件下衍生10 分钟,结果显示温度在60℃条件下时衍生化产物的色谱响应值较高,结果见表2,因此,采用在60℃条件下衍生10 分钟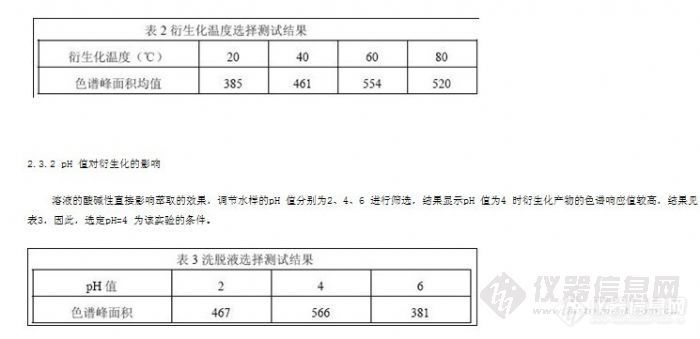
2.4 DC 衍生化产物标准曲线的制作
配制一组二苯氰胂标准溶液,浓度分别为0.005、0.01、0.02、0.03、0.04mg/L,在上述选定的条件下进行测定,衍生产物峰面积与二苯氰胂含量呈线性关系。校正曲线为Y=15.221X-0.4462,相关系数r=0.9996。
2.5 方法的回收率、精密度和检出限
按以上方法,用二苯氰胂溶液进行加标回收试验,回收率在95.3%~104.2%之间。配制同一浓度样品6 份,按照以上试验方法进行测试,计算色谱峰面积的相对标准偏差为2.9%,以噪音的3 倍响应为检出限,其检出限为20pg。
3 结 论
研究了水中二苯氰胂的固相萃取衍生化气相色谱的检测方法,选取了衍生化条件,固相萃取条件,定量测定最低浓度可达5.0×10-6g/L,最小检出限为20pg
参考文献
[1] 梁汉昌. 痕量物质分析气相色谱法. 北京: 中国石化出版社, 2000.
[2] 王立,汪正范,牟世芬等. 色谱分析样品处理[M]. 北京: 化学工业出版社,2001. 84~94.g