-
+关注
私聊
-

yhl-87_
第71楼2009/11/06
农药残留检验方法系列讲座(56)加速溶剂萃取/凝胶渗透色谱-固相萃取净化/GC-MS
加速溶剂萃取/凝胶渗透色谱-固相萃取净化/GC-MS
测定茶叶中残留的33种农药
摘 要: 建立了茶叶中有机磷、有机氯、拟除虫菊酯类共33种农药残留的分析方法。样品以丙酮一二氯甲烷(体积比为1:1)为提取剂经加速溶剂方法萃取,提取液用凝胶渗透色谱净化除去大部分的色素、脂类和蜡质,再用Carb-NH2:小柱和Florisil小柱净化。采用气相色谱法分析、外标法定量、气相色谱-质谱法(GC-MS)定性。加标水平为0.05 mg/kg时,大部分农药的回收率为70%~120%,相对标准偏差小于20%。方法的检测限为0.005~0.05 m g/kg (以10倍信噪比计)。该方法的提取效率高,准确灵敏,目前已应用于出口茶叶中农药残留的日常检测。大量实际样品的检测结果表明,此方法适于出口茶叶中农药残留检测实际工作的需要。
本文由绍兴出入境检验检疫局的胡贝贞, 宋伟华, 谢丽萍, 邵铁锋等多位研究人员共同完成,他们将已应用于出口茶叶中农药残留的日常检测分析方法,特别是收集淋洗液的经验值得借鉴,现全文介绍如下,第一作者为通讯联系人 E –mail:hubeizhen001@ 163.com.
前 言
茶叶是我国传统的大宗出口商品,茶叶进口国如欧盟、日本等发达国家对进口茶叶制定了非常苛刻的农药最大残留限量标准(MRL )。近年来,茶叶中农药残留检测项目不断增加,最大残留限量大幅降低,限量标准日趋严格。为此,建立茶叶中多种类型的农药多残留分析方法具有极大的实际应用价值。
近年来 ,对茶叶中农药残留的分析方法有不少研究报道[1~8] ,其中气相色谱法(GC)[1~3] 和气相色谱-质谱联用法(GC-MS) [4~7] 以其简便、快速、灵敏等特点而成为最常用的检测方法。茶叶中农药残留提取的传统方法有超声法[1]、均质法[3]、振荡法[6]等,这些方法往往存在有机溶剂用量大、耗时长以及残留农药提取不完全的情况。而微波辅助萃取(MAE)[2]、固相微萃取(SPME)[2 ],搅拌棒吸附萃取(SBSE) [4]等新技术也已应用于茶叶中残留农药的提取,这些方法减少了溶剂消耗,缩短了操作时间。加速溶剂萃取(ASE)是一种新型的样品提取方法,对于固体样品中的有机化合物萃取具有提取效率高、时间短、所用溶剂少等特点,已被美国环保署(EPA)收录为处理固体样品的标准方法之一[9],并已应用于土壤[9]、食品[11]、药物[12]等样品中残留物的提取。张桃英〔13〕曾采用ASE提取了茶叶中残留的六六六和滴滴涕。目前,国内外未见应用ASE同时萃取茶叶中残留的有机磷、有机氯和拟除虫菊酯类农药的报道。
本文结合实际工作需要,根据欧盟、日本以及我国出具卫生证书所要求重点检测的茶叶中农药残留的检测项目,在参考已有文献的基础上,重点考察了茶叶中残留农药的提取和净化方式,建立了茶叶中有机磷、有机氯、拟除虫菊酯类共33种农药的分析方法。实验考察了ASE方法对茶叶中上述3类农药的提取效果,并与传统的超声波提取、均质提取方法进行了比较。结果表明,ASE能快速、有效、完全地提取残留的农药。实验中还采用凝胶渗透色谱(GPC)和固相萃取结合的方法对提取液进行净化处理,结果发现GPC能有效除去提取物中的大部分色素、脂类、蜡质等大分子杂质,Carb-NH2小柱和Florisil小柱能进一步除去剩余的色素和一些小分子干扰物,净化效果很好。本方法采用GC-MS进行定性确证,采用气相色谱-火焰光度检测法(GC-FPD)和气相色谱一电子捕获检测法(GC-ECD)分别对有机磷、有机氯和拟除虫菊酯类农药进行定量分析。方法准确、灵敏、简便,且各项指标均能满足目前我国农药残留监测和发达国家的MRL要求。
实 验
1 实验部分
1.1 仪器与试剂
Agilent 6890N GC(配FPD和ECD检测器),Agilent 5975B GC-MSD(美国Agilent公司);ASE-300快速溶剂萃取仪(美国Dionex公司);凝胶渗透色谱仪(德国LC-Tech公司);固相萃取装置(美国Supelco公司);旋转蒸发仪(上海亚荣生化仪器厂)。Carb- NH2小柱:500mg/500mg,6mL (杭州福裕科技服务有限公司);Florisil小柱:1000mg,6mL(杭州福裕科技服务有限公司);丙酮、二氯甲烷为色谱纯;环己烷、乙酸乙醋为农残级;33种农药(具体名称见表1)的标准品(纯度大于99%,德国Dr.Ehrenstorfer公司)。
标准储备液的制备:准确称取10mg (精确到0. 1 mg) 农药各标准品分别于25 mL容量瓶中,用乙酸乙醋一环己烷 (体积比为1:1) 溶解,配制成400μg/mL的单标储备液,于-18℃条件下储存。
1.2 样品前处理
1.2.1 ASE提取
称取 4. 0 g 茶叶试样 (粉碎成40-60目),移人加速溶剂萃取仪的34 mL萃取池中,用丙酮一二氯甲烷 (体积比为1:1,以下简称溶剂A)作为提取溶剂,在10.34 MPa (1500psi) 压力、100℃条件下,加热5min,静态萃取10 min。循环1次。然后用池体积30%的溶剂A冲洗萃取池,并用氮气吹扫100 s。萃取完毕,将萃取液转移到100 mL鸡心瓶中,于35℃水浴中旋转蒸发近干,然后用适量乙酸乙醋-环己烷 (体积比为1:1,以下简称溶剂B) 溶解残余物后转移至10 mL离心管中,再用溶剂B定容至10mL。将这10mL待净化液高速离心后过0.45μm滤膜,供凝胶色谱净化用。
1.2.2 凝胶色谱净化
净化柱:500 mm ×25 mm,填装50g BioBeads-X3填料 (中性、多孔的聚苯乙烯一二乙烯基苯微球体;排斥极限:相对分子质量400~14000),柱床高32 cm;流动相:乙酸乙醋一环己烷(体积比为1:1);流速:5.0 mL/min;样品定量环:5.0 mL;预淋洗体积:85 mL;洗脱体积:50 mL;清洗体积:35 mL。取“1.2.1”节中所得到的5 mL待净化液按上述凝胶色谱条件进行净化,收集洗脱液于100 mL浓缩瓶中。
1.2.3 固相萃取净化
取Carb- NH2小柱,用5mL溶剂A预淋洗,保持该柱湿润,备用。将“1.2.2”节中收集的50 mL洗脱液转移到100 mL鸡心瓶中,于40℃水浴中旋转蒸发至约1 mL,移至Carb-NH2小柱上,再用2 mL溶剂B分两次洗涤鸡心瓶,将洗涤液一并加到该柱上。待净化液通过该柱后,用15 mL溶剂A洗脱。收集的洗脱液于35℃水浴中旋转蒸发至近干,加2 mL溶剂B溶解并定容。将该溶液平均分成2份,1份用于GC-FPD测定有机磷类农药,另1份用于GC-ECD测定有机氯和菊酯类农药。在GC-ECD测定中,取Florisil小柱,先用5 mL乙酸乙酯-环己烷 (体积比为5: 95 ) 预淋洗,然后加入上述1份 (1.0 m L) 溶液过柱,用10m L乙酸乙酷-环己烷(体积比为5:9 5 )洗脱,洗脱液于40℃水浴中旋转蒸发至干,加人1.0 m L环己烷定容后用于GC-ECD测定。
1.3 定性和定量方法
本实验采用GC-MS作定性确证;有机磷类农药用GC-FPD定量,有机氯和菊酯类农药用GC-ECD定量。
1.3 .1 GC-MS条件
色谱柱:HP-5 ms石英毛细管柱30m×0 .25 mm ×0 .25 μm。柱温:60℃(1min),30℃/min 升至200℃(1 min)再以15℃/min升至280℃(7min)。载气:氦气,纯度为99.999%,流速1.0 mL/min。进样口温度:260℃。进样量:1μL。进样方式:不分流进样,2.0min后打开分流阀。采用电子轰击离子源(El),电子能量70 eV。离子源温度:230 ℃。接口温度:280℃。采用选择离子监测(Sim)方式:每种化合物分别选择3~4个定性离子(见表1)。

-
+关注
私聊
-
yhl-87_
第72楼2009/11/06
农药残留检验方法系列讲座(56)(下)加速溶剂萃取/凝胶渗透色谱-固相萃取净化/GC-MS
1.3.2 GC-FPD条件
色谱柱:HP-5石英毛细管柱30m×0 .32 mm ×0 .25 μm。柱温:100℃(1min),以30℃/min 升至180℃再以10℃/min升至200℃(1min),再以30℃/min升至230℃(5.5min)。载气:氮气,纯度为99.999%。进样口温度:180℃。进样量:1μL。进样方式:恒压模式,138kPa ( 20.0 psi); 不分流进样,0.75 min后打开分流阀。检测器温度:240℃;辅助加热区温度:240℃;氢气流速:75 mL/min;空气流速:100 mL/min;尾吹气:氮气,流速60 mL/min.
1.3.3 GC-ECD条件
色谱柱 : HP-5石英毛细管柱,30m×0 .32 mm ×0 .25 μm。柱温:80℃(1min),以25℃/min 升至220℃(1min),再以6℃/min升至250℃,再以2℃/min升至260℃,再以6℃/min升至290℃(0.5min)。载气:氮气,纯度为99.999%。进样口温度:260℃。进样量:1μL。进样方式:恒压模式,1.5mL/min; 不分流进样,0.75 min后打开分流阀。检测器温度:300℃;尾吹气:氮气,流速60 mL/min.
2 结果与讨论
2.1 提取方式的选择
本实验选用适合各类农药提取的二元混合溶剂丙酮一二氯甲烷(体积比为1:1)作提取溶剂,并比较了ASE、超声波、均质3种提取方式。由于未能获得含有全部33种农药的阳性样品,实验中选取了一个同时含有水胺硫磷、三唑磷、氯氰菊酯、氰戊菊酯4种农药的阳性样品,按3种提取方式分别提取。ASE条件见“1.2.1”节所述。超声波提取条件:取1g粉碎过的茶叶,加5 mL水浸泡2h,再用30 mL丙酮一二氯甲烷(体积比为1:1)超声提取15 min;将残渣重复提取一次。均质提取条件:取1g粉碎过的茶叶,加5 mL水浸泡2h,再用30 mL丙酮一二氯甲烷(体积比为1:1)均质提取1 min;将残渣重复提取一次。3种提取方式的检测结果见图1。
从图1可见,ASE的提取效率高于超声波提取法和均质提取法。为了确保提取效率,保证检测结果的准确性,本方法选用ASE为提取方式。在“1.2.1”节所述的条件下,对阳性样品一次提取后的残渣重复提取一遍,未检出上述4种农药,说明在本实验选用的ASE条件下能完全提取残留在茶叶中的农药。
2.2 净化方式的选择
2.2.1 GPC净化
GPC净化能有效地去除样品中的脂类、色素等大分子干扰物,常用于脂肪含量较高的动物源性食品的净化[14],但很少有文献报道将GPC应用于茶叶样品的净化。茶叶基体复杂,其中含有大量的色素、蜡质等大分子物质,而使用ASE萃取更会促使大量的这些杂质被共提取出来。本实验考察了GPC对茶叶样品的净化效果。GPC切割时间点的选择要充分考虑被渗透排阻的干扰组分和目标化合物的分离情况。
本实验按以下条件做流出组分的收集:前运行17 min,弃去;最后运行5 min,弃去;主运行:收集第18,19,20,21~27,28~30,31~32 min共6个时间段的流出组分。结果发现:色素完全流出的时间为20 min,大部分有机磷、有机氯类农药组分集中在第21~27 min流出,水胺硫磷在第20min有部分流出,菊酯类农药组分由于其相对分子质量较大,在第18 - 20 min有相当一部分已流出,而λ-氯氟氰菊酯和氟氰戊菊酯则在18 min之前已有部分流出,在28 min以后各种农药已基本无流出。综合考虑净化效果和农药的回收率,确定收集的时间段为第18~27 min共50 mL流出物。
2.2.2 固相萃取净化
茶叶中含有大量的叶绿素、红茶色素、胡萝卜素、茶多酚和其他大量不确定的干扰物,因此尽管采用GPC净化已除去大部分的色素等大分子杂质,但还需做进一步的净化处理以除去剩余的色素和其他小分子杂质。活性炭小柱能很好地吸附色素,氨基柱能有效地吸附有机酸和糖分,将这两种小柱串联在一起净化效果较好。本实验比较了丙酮一二氯甲烷(体积比为1:1)和乙睛一甲苯(体积比为3:1)两种洗脱溶剂,发现用15mL这两种溶剂均能将目标物洗脱下来,但丙酮一二氯甲烷沸点较低,后续浓缩操作较方便,故选用丙酮一二氯甲烷作洗脱剂。净化液过Carb-NH2小柱后可供GC-MS作定性确证或供GC-FPD分析有机磷类农药。在进行有机氯和菊酯类农药分析时,因ECD检测器较易受污染,故净化液需预先过弗罗里硅土(Florisil)柱 除去一些含电负性杂原子的极性干扰物,再进行ECD测定。
2.3 方法的准确度和精密度
取阴性茶叶样品作0.05mg/kg浓度水平的添加回收试验,重复5个平行实验,计算各种农药的回收率和相对标准偏差(RSD),以考察方法的准确度和精密度,结果见表1。从表 1 可见,除了敌敌畏和λ-氯氟氰菊酯外,其余农药的回收率均在70%~120%之间且RSD≤10.10%。敌敌畏回收率偏低应该是其沸点较低,减压浓缩过程损失较大引起的。λ-氯氟氰菊酯回收率偏低是由于其在GPC上流出较早,在前运行阶段被弃去一部分所致。
2.4 线性范围和检测限
准确配制质量浓度分别为0.005、0.01、0.05、0.1、0.5、1. 0μg/mL的标准溶液,依次按“1.3.2"节所述和“1.3.3”节所述的条件进行分析,以峰面积Y对质量浓度X (μg/mL)作图。各农药检测的线性范围、线性方程及相关系数见表1
本方法的检测限根据10倍信噪比计算得出,各农药检测限结果见表1。各农药的检测限均能满足日本、欧盟等国家规定的最大残留限量要求。
3 实际样品测定
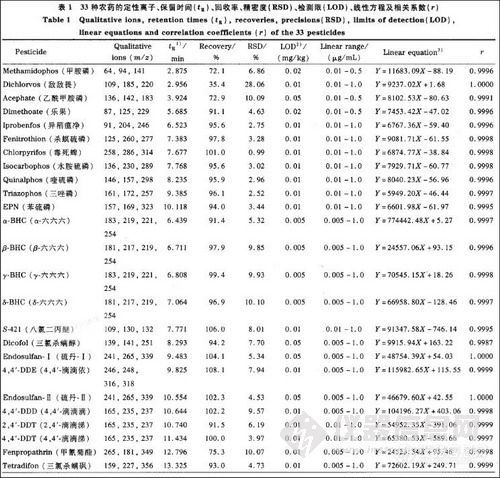
利用所建立的方法对实际样品进行测定。图2-a,b 分别是0.1 R ,g/mL有机磷农药标准溶液和一阳性样品的GC-FPD谱图。图3-a, b分别是0.1μg/mL有机氯及菊酯类农药标准溶液、与图2-b同一阳性样品的GC-ECD谱图。在这一阳性样品中检出的农药为毒死蝉、水胺硫磷、三唑磷、硫丹(I十II),氯氰菊酯、氰戊菊酯,其质量浓度分别为0.014、0.025、0. 101、0. 158、0. 175和0.323 mg/kg。33种农药标准溶液(0.1μg/mL)以及与图2-b、图3-b同一阳性样品的GC-MS SIM 谱图分别见图4-a,b。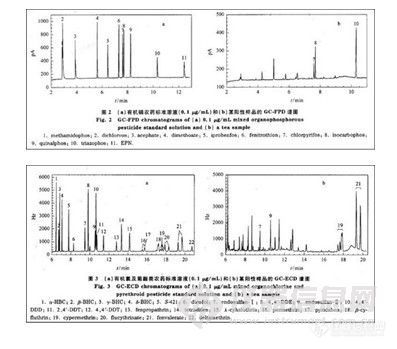

4 结论
本文建立了加速溶剂萃取、凝胶渗透色谱结合固相萃取净化测定茶叶中有机磷、有机氯、拟除虫菊酯3类共33种农药残留的方法。方法灵敏度能满足国外最大残留限量的要求;准确度和精密度高,在0.05 mg/kg添加水平时,大部分农药回收率处于70%和120%之间,RSD≤10.10%。方法的提取效率高,净化效果好,可用于进出口茶叶中农药残留的日常检测工作。

-
+关注
私聊
-
yhl-87_
第73楼2009/11/06
农药残留检验方法系列讲座(57)有机氯农药残留分析方法标准化的研究
摘 要:概述了农药残留分析方法国家标准制订中在方法验证、分析步骤、条件对比试验、干扰及分离度研究、多个实验室质量控制等内容,为方法标准化提供可资借鉴的实例。
本文由农业部环境保护科研监测所的黄士忠、陈国光、王继军完成,发表在《农业环境与发展》上的论文,向大家介绍他们是怎样制定标准化方法的过程,对农残分析工作者有参考价值。现全文介绍如下。
前 言
我国自50年代初广泛、大量使用BH,DDT至1982年停产、停用,累计使用BHC原粉400万吨,DDT原粉50万吨,平均每亩耕地投放:BHC 2~3公斤、DDT 0.5公斤,占农药总用量的50%。由于这两种农药化学性质稳定,不易分解,在土壤中消失需要20年之久,并通过食物链向生物及人体内转移、蓄积。虽然停用已近12年,但近年来调查发现,土壤及农畜产品中污染普遍存在,以至BHC, DDT残留量仍是我国农副产品进出口的必测项目。因此,制订土壤、生物中BHC, DDT分析方法国家标准是势在必行。因为它在探索环境生态系中的浸污程度净化效应、转移、变迁、演变、归宿规律、判断和预测农业环境质量、评价防治措施及效果等方面,在环境检测、外贸出口、商检、法检及卫生检验等部门,都有广泛的用途。
1 样品提取方法的对比试验
土壤、藕、猪肉、鱼和鹌鹑中六六六、DDT气相色谱的提取方法对比试验。
为使制订出的六六六、DDT农药残留分析方法更具代表性和可行性,因而本文拟以土壤、样品中BHC, DDT残留量气相色谱法分析中提取方法对比试验为例,在除提取方法外其它试验条件完全相同的情况下,比较试验结果,从而选择最合适提取方法(试验按GB/T 1450-93和GB/T1451-93的分析步骤进行),结果见表1。
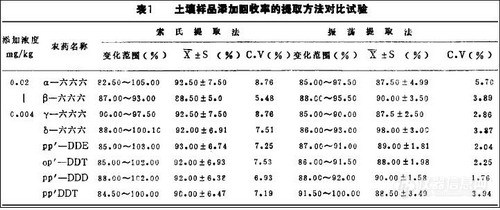
从表中所示的结果看,两种提取方法都可以作为标准的提取方法。可根据样品量的多少选用。
2 不同净化方法的对比试验
采用鱼样品中BHC, DDT残留量的色谱分析中,在提取及检测条件完全相同的情况下,比较浓硫酸净化法和酸性硅藻土柱净化法的试验结果见表2。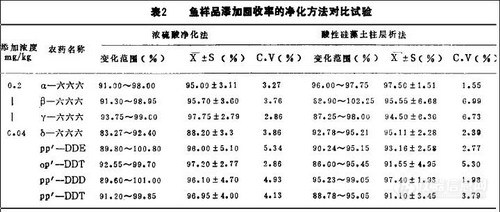
从表2的结果看,鱼样品采用浓硫酸净化法的平均回收率为:89.2~95.6%,38.2~97.75%,变异系数分别为2.32~-5.36%; 2.86~-5.34%。而采用酸性硅藻土柱层析法的平均回收率分别为91.5~-99. 35%, 91.1~-97.3 %,变异系数分别为0.97~4.49%, 1.55~6.99%。表明这两种净化法都可以作为标准的净化法。
3 干扰影响的研究
目前国内外检测BHC, DDT等有机氯农药残留量的色谱柱,一直沿用固定液为1.5%OV一17 + 1.95%QF- 1的色谱柱,该柱子的优点是能在20min内很好地分离出BHC, DDT八种异构体或衍生物,分离度好、检测灵敏度高,是其它色谱柱所不及的。但是,在所规定的色谱检测条件下,七氯会干扰β-BHC、狄氏剂会干扰pp-DDE的测定,所以在检测时很容易造成卜BHC和pp-DDE残留量的错误结果。通过降低气流的办法可以基本了解样品中是否含有七氯和狄氏剂,见色谱图1。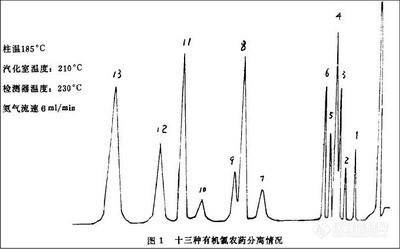
如果发现有七氯和狄氏剂,可改用下列色谱柱来检测β-BHC和pp -DDE;色谱柱5%OV-210/Chromosorb WHP80~ 100目,柱长200cm,内径4 mm。
汽化室温度230 ℃,柱温200℃,检测器250℃,氮气流速50m1/min。
4 方法的确证定性试验
为了防止一根色谱柱上出现具有相同保留值的其他物质,保证测定物质的准确性,
可采用两根色谱柱进行确证定性试验。
1. BHC. DDT及主要异构体、代谢物(8种化合物)的标准溶液采用1.5%OV-17+1.95
QF-1/Chromosorb W AW-DMCS和80~100目色谱柱,在柱温202℃,汽化室220 ℃,检定器245℃的条件下检测,其色谱图见图2。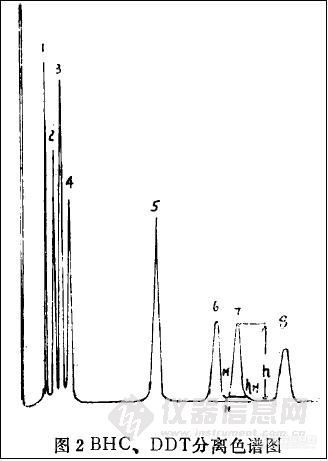
2. 为了确证BHC, DDT及主要异构体、代谢物(8种化合物)标准样品,在采用上述气谱柱和工作条件测得图谱和保留时间后,又采用1.5%OV-17+1.95OV-210/chromosorb W AW- DMCS, 80~100目,柱温204℃,汽化室220℃,检定器245℃的条件,所测图谱见GB/T 14551-93,第6页,这里从略。
用上述2根色谱柱测定同一农药标准溶液,都含有8种化合物,因温度和固定液有所不同,其保留时间表3所示有差异,但峰形、分离情况、峰高等基本相近。由此确认是所需要测定的BHC,DDT的8种化合物。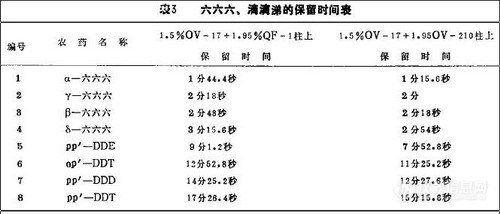
5 色谱柱对BHC和DDT分离度的影响
分离度是评价柱效及选择性的总指标。本方法采用1.5%OV-17+1.95%QF-1/chromosorb W AW-DMCS 80~100目和1.5%O V-17
+1 .95%OV-210/chromosorb W AW -DMCS80~-100目,两根色谱柱对BHC和DDT的8种化合物的分离度,都达到方法规定的要求。
我们采用标准溶液在这两根柱上进行了分离效果试验。
1. 在1.5% OV-17+1.95%QF-1/chromosorb W AW-DMCS 80~100目柱,柱温204℃,汽化室220℃,检定器245℃工作条件下,其分离度见图2
分离度: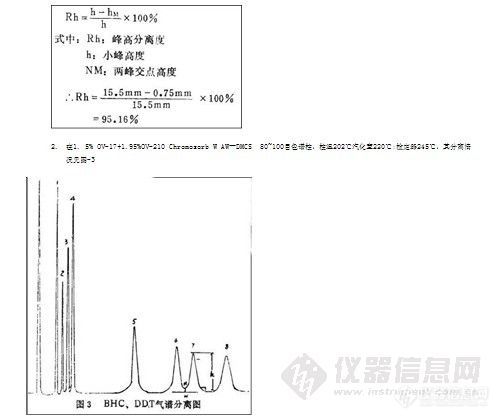

从上述两根色谱柱的分离效果看,虽然OP-DDT和PP-DDD是这8种化合物中最难分离的2种,但这二根柱上的分离度分别达到95.16%和93.82%,都达到了较好的分离水平,其它化合物的分离度都达到96 %以上,完全满足标准的要求。
6 方法试验的质量控制
统一分发样品,按照《农药残留分析方法初探》(以下简称《初探》)中提出的要求,制定允许差,并建立精密度和准确度质量控制图。
1. 允许差 本方法进行室内允许差和室间允许差的计算。粮食(玉米)中六六六、DDT的浓度为0.2~-0.04ppm。按上述“初探”中的公式进行计算,允许差的结果见表4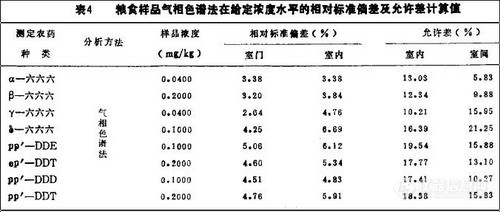
2. 精密度质量控制图每对数据之间极差Ri单位mg/kg,实测结果见图4、5。用气相色谱法分析粮食(玉米)中六六六、DDT加标百分回收率,得出其准确度质量控制图如图6, 7所示。
从上述精密度和准确度质量控制图看,在我们协作试验的粮食中,六六六、DDT分析的精密度和准确度都在控制范围内。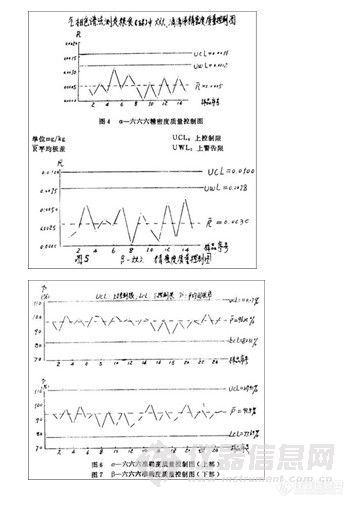
7分析方法的评价
精密度及准确度的结果(参看GB/T14551-93,第8页)
方法的验证采用GB/T 14551-93,测定玉米中六六六和DDT。方法灵敏度、准
确度和精密度是评价的主要技术指标,现将试验结果归纳如下。
1.方法灵敏度,是指可供分析数据的最低样品浓度,以检测极限表示。玉米样品最小检测浓度的变化范围为0.000049~0.00487mg/kg 。允许残留量(mg/kg) 粮食中六六六为0.5滴滴涕为0.1所以方法的最小检测浓度比允许残留量高2~4个数量级。
2.方法准确度,用添加回收率来表示。其变化范围为90.4~-98.2%。
3.方法精密度,通常用标准偏差或变异系数表示。变化范围为1. 02~12.73% .上述三个指标和规定的要求比较,均符合农药残留分析有关规定。
以上七个方面的概述,较详细地分析、介绍了农药残留分析方法标准制定的全过程
和操作方法,望能对以后农药残留分析方法标准的制订有所帮助。
参 .考 文 献
1黄士忠土城质t六六六和滴滴涕的侧定气相色谱法,北京,标准技术出版社,1994, 14550
2货士忠生物质量六六六和滴摘涕的测定气嫩j色谱法,北京,标准技术出版社,1994, 14551
-
+关注
私聊
-
yhl-87_
第74楼2009/11/06
农药残留检验方法系列讲座(58)-牛乳中多种有机磷农药残留的双色谱柱法
摘 要 牛乳中的有机磷农药残留经乙酸乙酯萃取后浓缩进样用双毛细管柱(DB-1 、DB-17) .双火焰光度检测器FPDl、FPD2)测定牛乳中13种有机磷农药残留测定结果回收率72.38%~102.7%,相对标准偏差1. 37%一7. 21 %,方法检出限为0.005~0. 012 mg/ kg
本文由云南省农业科学院质录标准与检测技术研究所,农业部昆明农产品质录监督检验测试中心的梅文泉、黎其万和饵注等研究人员发表在《食品与发酵工业》杂志上的论文,现全文转发给大家参考
前 言
牛乳中有机磷农药残留含量是影响牛乳质量安全的因素之一。在中华人民共和国农业行业标准《无公害食品》中规定.牛乳中甲胺磷、倍硫磷、久效磷、甲拌磷、杀扑磷等有机磷农药残留不能超标[1]。及时、快速、准确测定牛乳中有机磷农药残留.对保证牛乳质量.防止有机磷农药残留超标的牛乳上市销售、保障人民身体健康具有重要意义。有关测定牛乳中有机磷农药残留的方法有文献报道[2~4]但这些方法有的试剂用量多、花费时间长、有的测定有机磷农药种类少,,而且均用单柱、单检测器进行测定,不易准确定性。本试验采用乙酸乙酯萃取.三阶程序升温、双石英毛细管柱分离.双火焰光度检测器测定牛乳中13种有机磷农药残留.测定结果准确度高.重现性好.测定有机磷农药种类多.操作简单、快速.满足测定牛乳中多种有机磷农药残留的需要。
实 验
1 材料与方法
1. 1仪器与试剂
美国Agilent 6890N型气相色谱仪.带双火焰光度检测器 (FPD1和FPD2.磷滤光片).分别装有非极性石英毛细管柱DB-1( 30 m×0. 53 mm×1. 5 um)和中极性石英毛细管柱DB-17 ( 30 m × 0. 53 mm × 1. 0um.配备双塔自动进样器、化学工作站;德国4000型旋转蒸发仪;国产HY-2调速多用振荡器。
乙酸乙酯、丙酮、无水硫酸钠均为分析纯.乙酸乙酯和丙酮重蒸.无水硫酸钠在 140℃烘箱中烘6h。
农药标准品敌敌畏、甲胺磷、乙酰甲胺磷、甲拌磷、氧化乐果、久效磷、乐果、甲基对硫磷、毒死蜱、对硫磷、倍硫磷、喹硫磷、杀扑磷等13种有机磷农药标准品购于中国标准技术开发公司。
1. 2气相色谱条件 进样口温度220 ℃.检测器温度250 ℃;柱温采用二阶升温程序即150 ℃保持2 min.以8℃/min升温至200℃.保持5min.以10℃/min升温至250℃.保持10 min;载气为N2.流速为10 m1/ min; H2 75 m1/min;空气100 m1./ min。不分流进样.进样量1 μL。以双柱保留时间定性.以DB-17获得的峰而积外标法定量。
1. 3试验方法
称取市售均匀灭菌牛乳5. 00 g,置于100 m1.具塞锥形瓶中.加入25 mL乙酸乙酯.振荡器上振荡提取20 min.加入l0g无水Na2S04.再振荡提取10min.倒入铺有滤纸的漏斗中过滤.锥形瓶用10 m1.乙酸乙酯分2次洗净.滤渣用10 m1.乙酸乙酯分2次冲洗.集中滤液于150 m1.浓缩瓶中.将浓缩瓶置于真空旋转蒸发器上.在35 ℃温度下浓缩至2~3 m1,.将浓缩液转移到刻度离心管中.准确定容到5. 0 m1。在旋涡混合器上混匀后.倒入气相色谱专用样品瓶中上机同时用双毛细管柱、双检测器进行测定。
2 结果与分析
2. 1色谱分离结果
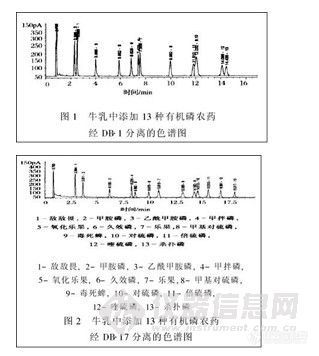
将13种农药标准按一定比例添加到牛乳样品中.按1. 3提取后测定的牛乳中13种有机磷农药经DB-1 ,DB-17分离.其色谱图见图1、图2。
2. 2有机磷农药的双柱、双检测器定性
有机磷农药种类繁多.用一根柱子进行分离测定时.会有2种或2种以上的有机磷农药具有相同的保留时间.用单柱单检测器对这些农药定性不准确。本试验用极性不同的2根毛细管柱进行分离.双检测器同时测定可提高农药检测的准确度。例如.毒死蜱、对硫磷和倍硫磷在DB-1上的保留时间十分接近.无论怎样调整色谱条件都无法分开(见图1) ,而采用中极性柱DB-17可以使这3种农药得到较好的分离(见图2)。
2. 3 十三种有机磷农药保留时间、线性回归方程、相关系数及方法检出限测定结果
配制单一有机磷标准溶液.分别进样确定各种有机磷农药的保留时间。
配制浓度为0. 02 ,0. 05 ,0. 10 ,0. 5 ,2. 0 μg/ m1,的13种有机磷农药标准溶液。气相色谱分析结果以DB-17获得的峰面积(Pa* S)对标准浓度(μg/m1,)进行线性回归.得到线性回归方程和相关系数在样品中加入13种农药混合标准溶液.使样品中添加农药标准的浓度为0. 01 mg/ kg,然后按1. 4条件处理样品.以DB-17获得的色谱图按信噪比3倍条件计算方法检出限。
13种有机磷农药保留时间、线性回归方程、相关系数及方法检出限测定结果见表1。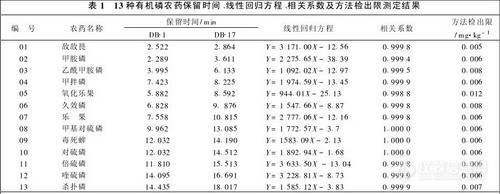
2. 4 牛乳中13种有机磷农药回收率测定结果
在牛乳样品中添加13种有机磷标准工作液.添加后样品中13种有机磷农药浓度为0. 10 mg/ kg,混匀放置0. 5 h后按1. 3步骤进行测定。做3次重复.结果见表2。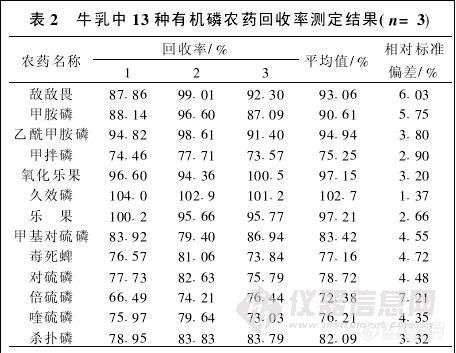
2. 5 本文方法与GB/ T 17331-1998方法的比较
在中华人民共和国农业行业标准《无公害食品》中.规定测定牛乳中甲胺磷、倍硫磷、久效磷、甲拌磷、杀扑磷等有机磷农药残留量需采用G B/ T 17331一1998 [2]。笔者就本文方法与G B/ T 17331-1998方法进行了比较试验.比较试验结果见表3

本文方法采用对含磷化合物具有高选择性的火焰光度检测器,大大简化了净化步骤,使各种有机磷农药的损失最小,13种有机磷回收率72..38%~102.7% (见表2)。GB/ T 17331-1998方法由于操作步骤多,甲胺磷等有机磷农药在提取和净化过程中损失严重。笔者采用GB/ T 17331-1998方法对甲胺磷的回收率进行多次测定.回收率在30%以下 ( GB/ T 17331-1998测定范围不包含甲胺磷[2] )。本文方法测定多种有机磷有较好的回收率,但由于未对样品进行净化,牛乳中的脂肪和杂质可能会聚集在毛细管柱的前端而影响其柱效和寿命,可以采取两项措施延长柱子寿命,一是在进样口下的衬管中放置玻璃棉,用玻璃棉滤除样液中的部分脂肪和杂质.做50个左右的样品后更换玻璃棉;一是在柱子的前端加一段1 m长的空毛细管柱,污染后及时更换。
由表2可以看出,回收率为72. 38%~102.7% ,变异系数为1. 37%~7. 21%,说明该试验方法具有较好的准确度和精确度。
3 讨论
本试验采用乙酸乙酯提取,三阶程序升温,双石英毛细管柱分离,双火焰光度检测器同时测定牛乳中13种有机磷农药残留。标准曲线线性关系良好,方法检出限、回收率、变异系数符合农药残留指标中灵敏度、准确度和精确度的要求[5]。本文试验方法定性准确、测定农药种类多、重现性好,分析速度快,操作简便、快速,满足测定牛乳中多种有机磷农药残留的需要
-
+关注
私聊
-
yhl-87_
第75楼2009/11/06
农药残留检验方法系列讲座(59)蜂蜜中多种有机磷农药残留量的气相色谱测定
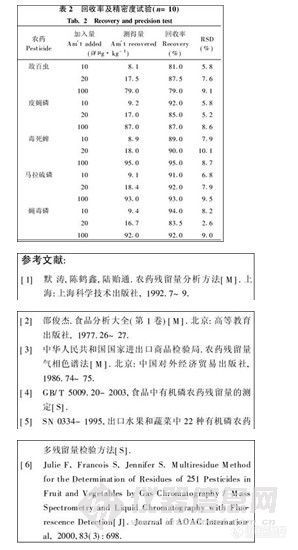
摘 要:介绍了应用配有FPD检测器的气相色谱仪测定蜂蜜中多种有机磷农药残留量的新方法。试样加水稀释.加入少量氯化钠.再用乙酸乙酯提取。以GC-FPD测定.外标法定量.必要时结果用不同极性的毛细管柱验证分析结果。敌百虫、皮蝇磷、毒死蜱、马拉硫磷和蝇毒磷分别在一定范围内,质量浓度与峰面积呈良好的线性关系。方法准确,重现性、精密度好,杂质干扰少。方法的检出限均为0. 01 mg·k g-1。
本文由浙江出入境检验检疫局的朱青青、谢文和丁慧瑛等研究人员完成,注意本文是含糖粘稠样品,作者的前处理方法简单效果不错,值得参考。摘自《理化检验.化学分册》杂志第41卷。
前 言
蜂蜜是我国的传统出口商品,由于蜜蜂在采蜜过程中采集了被农药污染的花粉及蜂农使用了有机磷农药,导致蜂蜜中的有机磷农药残留量有可能超标。欧盟在对我国出口蜂蜜的主要安全卫生检测项目中就有对有机磷的检测要求。根据进口国的要求,我国制定残留监控计划要求检测蜂蜜中的多种有机磷.包括敌百虫、皮蝇磷、毒死蜱、马拉硫磷和蝇毒磷.,并且规定了严格的最高残留限量,敌百虫为0. 01 mg·k g-1、皮蝇磷为0. 02 mg·k g-1、毒死蜱为0. 02 mg·kg-1、马拉硫磷为0. 0l mg·k g-1和蝇毒磷为0. 02 mg·k g-1。虽然我国的残留监控计划己实施了二年但一直没有测定蜂蜜中多种有机磷的国家和行业标准.现有测定蔬菜、水果、粮谷中多种有机磷的国家标准和行业标准.不适用测定蜂蜜样品,因此都是采用各实验室的非标方法完成前二年的残留监控计划。
皮蝇磷、毒死蜱、蝇毒磷、敌百虫和马拉硫磷为有机磷类杀虫剂、杀螨剂[1],其性质不太稳定,同时由于蜂蜜粘稠,较蔬菜、水果、粮谷等样品前处理难度大。本文用GC-FPD测定蜂蜜中多种有机磷农药残留量可满足实际检验工作的需要。
实 验
1试验部分
1. 1仪器与试剂
H P- 6890气相色谱仪.配有FPD检测器。
敌百虫、皮蝇磷、毒死蜱、马拉硫磷和蝇毒磷标准溶液:称取皮蝇磷、毒死蜱、马拉硫磷和蝇毒磷标准品10 mg分别置于100 mL容量瓶中,加乙酸乙酯制成标准储备液。根据需要用乙酸乙酯稀释至适当浓度的标准工作液。水为蒸馏水,乙酸乙酯为色谱纯。
1. 2分析步骤
称取试样10. 00 g于50 m1.具塞塑料离心管中,加水15m1.和氯化钠5 g.混匀,加乙酸乙酯15 m 1..在混匀器上充分混匀2 min. 2 000 r· min-1离心3 min.用尖嘴吸管将上层乙酸乙酯溶液移至100 m1.浓缩瓶中,残渣加乙酸乙酯15m 1,重复上述操作,合步{两次提取液.40℃以下水浴减压浓缩至近干,平缓氮气流下吹至干,加入乙酸乙酯1 m1..混匀.溶液通过0. 45 u m滤膜过滤后.供GC测定。
1. 3色谱测定条件
DT3-1701石英毛细管柱(30 m/0. 25 m m/0.25 μm);载气氮气(>99. 999%)流量1. 0 m1·min-1;空气流量100 m1·m in-1;氢气流量75 m1·min-1。柱温:50℃(保持2 min)} 以15℃/min升到180℃(保持lmin),再以8/min上升到290℃(保持10 min).,进样口温度250℃;检测器温度250℃;尾吹氮气.30 m1·min-1,不分流进样.进样量2uL。
按仪器条件.对标准工作液和样液等体积穿插进样。根据峰面积用外标法定量。
2结果与讨论
2. 1提取溶剂的选择
乙腈具有极性大,穿透力强的特点[2,3],文献[4~6]选用二氯甲烷、乙酸乙酯和乙腈作为食品中有机磷农药的提取溶剂。经试验,乙酸乙酯和乙腈对各种有机磷农药都有很好的提取效率.考虑到乙腈的浓缩时要求的温度较高,敌百虫不稳定回收率会偏低,同时乙腈毒性大,本方法选用乙酸乙酯作为提取溶剂,从有机磷农药的测定结果看效果更佳。
2. 2方法的线性关系和最小检出量
在试验条件下将多种有机磷混合标准储备液稀释成5个不同的质量浓度,并且绘制标准曲线 (线性范围均为0. 1 ~1. 0 mg·L-1 ).线性方程和相关系数.见表1。在上述质量浓度范围内.多种有机磷农药的质量浓度与相应的峰面积值呈良好的线性关系。敌百虫、毒死蜱、蝇毒磷、皮蝇磷和马拉硫磷标准色谱图见图1。敌百虫、毒死蜱、蝇毒磷、皮蝇磷和马拉硫磷方法的检出限均为0.01mg ·kg- 1。

-
+关注
私聊
-
yhl-87_
第76楼2009/11/06
农药残留检验方法系列讲座(60)农药残留检测与样品前处理技术的发展趋势
一、概 述
农药是当前农业生产用于防治病、虫、杂草对农作物危害不可缺少的物质,对促进农业增产有极重要的作用。随着农业科学技术的发展,化学农药的品种和数量不断增加,已成为防治病虫害的主要手段。农药施用到农作物上以后,绝大部分因多种原因而转化,但作物内会残留有极少量的农药。长时间摄食残留农药会影响人体的健康,这就是农药残留量问题的由来。近年来,在茶叶、粮谷、蔬菜及水果种植中由于不少农户忽视农药的正确、合理使用,农药污染问题经常发生,农药残留量超标相当严重,并逐年加剧。而欧盟、美国、日本、加拿大等西方发达国家或地区,出于维护本国经济利益和保护人们健康的需要,相继对进口食品中农药残留量等卫生指标提出了愈来愈严格的要求。鉴于此,为保障我国人民的身体健康、有效控制农药在茶叶、粮谷、蔬菜和水果等生产中的合理使用和对其残留量进行监控,满足进出口贸易的需要,大力开展农药残留量检测技术以及相关的前处理技术的研究是非常必要的。
化学农药是一类复杂的有机化合物,根据其用途可以分为杀虫剂、杀菌剂、除草剂、植物生长调节剂、杀螨剂、杀鼠剂、杀线虫剂。根据化学结构又可分为有机氯、有机磷、拟除虫菊酯杀虫剂,取代氯苯氧基酸或酯除草剂,氨基甲酸酯杀虫剂、除草剂和杀菌剂和有机杂环类杀菌剂、除草剂等。农药残留量分析需要测定各种样品中ug/g、ng/g、甚至pg/g量级的农药和/或代谢产物及降解产物。其分析过程一般包括取样、样品处理(提取、净化和衍生化)和测量,根据农药种类和样品基质的不同,上述各个步骤的复杂性有所不同。色谱方法常用于样品的净化和测量,以前较多采用填充柱气相色谱法(GC),现在则越来越多地使用毛细管气相色谱法(GC)和高效液相色谱法(HPLC),尤其在定性分析的气相色谱/质谱法(GC/MS)中,毛细管柱技术占绝对优势。电子捕获检测器(ECD)、火焰光度检测器(FPD)、氮磷检测器(NPD)是最常用的农药残留量分析的气相色谱检测器,质谱检测器(MSD)则是最通用和灵敏的检测器。各种进样方式,如分流、不分流、冷柱上进样技术和程序升温汽化进样技术都已应用于农药残留物分析。近年来,随着农药残留研究的不断深入,农药残留检测方法日趋完善,并向简单、快速、灵敏、多残留、低成本、易推广的方向发展。
在检测技术方面,目前国际上已较多采用多残留检测技术和快速筛选检测技术
传统的农残分析大多用来分析某一类农药的单一成分,多残留分析方法(Multi-Residue Analysis Method)不仅可以用于分析同一类农药中的不同成分,而且可以分析不同种类农药中的不同成分。前者称为选择性多残留分析方法(Selective Multi-Residue Analysis Method),后者称为多类多残留分析方法(Multi-class Multi-Residue Analysis Method)。这方面,如英国中央科学实验室(CSL)开发了104种农药残留量同时检测的方法;德国科学研究协会开发了320种农药残留的多残留检测方法;美国FDA农药分析手册(PAM)的多残留方法可检测300多种农药;美国CDFA和荷兰卫生部都有较好的多残留同时检测方法和系统分析方法。这些方法,既可用于定量,又可进行确证。我国从上世纪90年代初开始研究和利用多残留分析方法,并相继推出了一系列国家标准。如国家标准GB/T17331-1998食品中有机磷和氨基甲酸酯类农药多种残留的测定和GB/T17332-1998食品中有机氯和拟除虫菊酯类农药残留的测定等,均可同时测定不同类型中的20多种农药残留。在我们的行业标准中SN/T0334-95多残留检测方法能同时检测22种农药残留量,秦皇岛局制定的《农产品中多种拟除虫菊酯残留量检验方法》已成为国际AOAC方法。在我局技术中心目前开发或采用的检测茶叶、蔬菜等样品中有机氯、有机磷、菊酯类农药以及一些杂环类农药的方法和本次研讨会将要学习和讨论的方法也大都是多残留检测方法。当然,我们现在的方法,在一次性检测农药的数量上和确证技术上与国际先进方法还存在不小的距离。
在快速筛选检测技术方面,上个世纪六十年代就有人利用薄层色谱酶抑制法测定有机磷农药等残留量,检测限量为毫克级;八十年代开始,农药的酶抑制和免疫检测技术作为快速筛选检测方法受到许多发达国家的高度重视,并因此得到了快速发展。酶抑制、酶联免疫(ELISA)、放射免疫(RIA)、单克降抗体等技术由于可以避免假阴性,适宜于阳性率较低的大量样品检测,在农兽药残留检测中应用日益增多。我国在近十多年来也相继开展了农药残留酶抑制法和免疫法快速筛选检测方法研究,取得了一定的研究成果,系统内有广东检验检疫局研制了农药残留速测卡,但总体上应用不多,方法的灵敏度不高,试剂不够稳定。
在样品前处理方面,现代色谱分析样品制备技术的发展趋势就是使处理样品的过程要简单、处理速度快、使用装置要小、引进的误差要小、对欲测定组分的选择性和回收率要高
目前,国际上较多使用固相萃取(SPE)、微波提取技术、凝胶层析(GPC)、加速溶剂提取(ASE)、基体分散固相萃取(MSPD)、超临界萃取(SFE)、固相微萃取技术。而我国目前主要采用传统的溶剂萃取,液液分配,柱层析净化,前处理方法自动化程度低、提取净化的效率不高,速度慢,环境污染严重。新开发的前处理技术其目的和结果就是要实现快速、有效、简单和自动化地完成分析样品制备过程。
下面就农药残留检测中采用的气相色谱检测技术和前处理技术的新发展向各位做一些简单的介绍。
二、检测技术
1、GC/MS 和GC/MS/MS技术
质谱技术问世于1910年。传统的有四极质谱仪和飞行质谱仪。近年来又出现了串联质谱仪(MS/MS)傅里叶变换离子回旋共振质谱仪等。目前,GC/MS的发展方向是小型化(即台式GC/MS)、自动化(仪器调试、控制、数据处理)、高灵敏度和高稳定性。目前几种常见的离子源有电子轰击型离子源(EI)、化学电离源(CI源)、快原子轰击电离源(FAB源)、解析化学电离源(DCI)、大气压电离源(API源)等。电子轰击型离子源(EI)是有机质谱应用最广的常规型离子源;化学电离源(CI源)又称软电离技术,可获得准分子离子峰,是EI源的一个补充;大气压电离源(API源)主要用作液相色谱-质谱联用。
(1)GC/MS技术
气相色谱测定农药残留的基本原理是根据保留时间来判定待测组分。往往因为样品提取和净化等原因,可能会出现许多杂质峰。如果待测组分在保留时间内有一种或多种杂质峰出现,就可能被认为是待测组分,造成误判。GC/MS法不仅根据样品中待测组分在图谱上的保留时间,更主要是根据在此保留时间内残留农药裂解的特征离子碎片,由质谱仪按其分子量和分子结构对农药准确定性,并以此作为定量的依据,从而克服了由于未净化掉的杂质峰与农药保留时间重叠而造成将杂质峰误判为农药的缺点。GC/MS技术在农药残留检测中已有许多成功的应用实例,在此不多作介绍。但随着农药残留限量要求的进一步提高,以及样品基质的影响,这种技术的应用也受到了一定的限制。大家切记,GC/MS方法的灵敏度不是由标准溶液的信噪比提供的,而是由样品基质条件下的信噪比决定的。
(2)GC/MS/MS技术
比一级质谱具有优势的是以离子阱为质量分析器的离子阱串联质谱仪具有与大质谱相当的功能,可提高灵敏度,可对复杂基体中微量待测物进行测定,对一级质谱无法区分的化合物可进行进一步的确认,以及同分异构体的区分。在分析领域,离子阱串联质谱已成为今后台式小型GC/MS的发展方向之一。有机磷农药在各种农作物和环境样品中含量很低,目前利用GC/MS技术分析农作物和环境样品中的农药残留量越来越普遍,因为它能给出化合物的结构信息,有利于化合物的定性。但因一般样品中(如蔬菜、水果、茶叶等)的背景干扰较大,导致样品预处理的周期较长,而且回收率较难保证。而MS/MS技术的应用,为复杂样品中微量农药的定性、定量分析提供了新的途径。在分析韭菜中倍硫磷农药时,分别采用EI全扫描和MS/MS分析,在MS/MS 分析条件下,倍硫磷的信噪比相对于EI全扫描时提高了100多倍。此外,利用MS/MS分析的另一大特点是可以将在色谱上不能完全分开的共流出物利用时间编程和多通道检测将其分开。
-
+关注
私聊
-
yhl-87_
第77楼2009/11/06
农药残留检验方法系列讲座(60)(2)农药残留检测与样品前处理技术的发展趋势
目前,GC/MS/MS已在环境分析、食品分析等方面得到广泛的应用。该技术不仅适用于复杂基体混合物的定性分析,而且可以利用得到二级质谱结果进行定量。这是因为在两个前后串联的质谱/质谱仪中,前级质谱主要用于担任分离工作,在样品被电离后,它只允许被分析的目标化合物的母离子或特征离子碎片通过,经过碰撞裂解后,再由第二级质谱分析裂解后产生的离子碎片,利用MS/MS可以同时得到较低的检测限和良好的结构鉴定信息(1个母离子和2个或更多的子离子)。与气相色谱检测器相比,传统的台式质谱仪(GC/MS)因灵敏度较低,其使用受到限制。而据文献报道,GC/MS/MS可在与传统气相色谱检测器相似的灵敏度下进行定性定量分析。原因是MS/MS在对离子检测前就排除了干扰,所以即使对复杂样本也可达到很高的灵敏度。它不需要重复进样就能定性,比选择性检测器有更高的可信性。在用传统的质谱仪分析较“脏”的样品时,因大量干扰的存在而不选择低于100amu的离子。因为许多样本的共存杂质含质量数低于100amu的离子;对检测器造成严重干扰,使被测物的碎片离子无法检出。在MS/MS中,子离子图谱中只有来自母离子的碎片离子,因此,低质量离子不受干扰,对结构鉴定更加有用。例如,建立乙酰甲胺磷分析方法时,即使质量数低至M/Z42,该离子由于不受干扰仍可作为定量分析离子。检测技术的发展已对残留量分析提出了更高的要求,即虽然传统中GC检测器可进行痕量分析并达到较低的检测限,但仍要求用MS作结构确证。因此任何实验室测定残留量的能力都会受到MS方法灵敏度的限制。选择离子检测(SIM)已被用于降低检测限。但是,SIM所收集的离子信息并不如全扫描丰富,不能认为是一个等同的结构分析。在大多数情况下,MS/MS的灵敏度相当于或低于GC选择性检测器的下限,并可在此水平上进行定量分析和真实的分子结构确证。美国纽约州农业署食品实验室已建立了一种用GC/MS/MS技术对水果、蔬菜和牛奶中100多种农药残留量进行检测、定量和结构确证的方法。这一方法可对浓度范围低至PPB水平的100多种农药进行准确的检测和鉴定。美国农业部的Beltsville农业研究中心利用GC/MS/MS技术分析了水果和蔬菜萃取物中22种农药残留物,得到良好的回收率和重现性,检出限小于2ng/g。当然,当前GC/MS/MS方法的局限性在于仅能检测目标分析物,很难一次进样分析大量化合物。王焕龙等报道了茶叶中有机氯农药残留量的气相色谱/串联质谱分析的研究报告,采用气相色谱串联质谱仪(GC/MS/MS)同时检测茶叶中51种有机氯农药。茶叶样品经二氯甲烷/水匀浆提取,用饱和氯化钠溶液分离水溶性色素杂质,再以弗罗里硅土(硅酸镁)固相萃取柱(SPE)净化,最后以GC/MS/MS检测分析,得到清晰的MS/MS质谱图,可去除茶叶背景值干扰,增加信噪比(S/N),适用于鉴定、确认分析极低浓度的定量分析。茶叶中色素相当多,样品经弗罗里硅土固相萃取柱(SPE)净化后,仍有色素杂质存在,会影响传统的GC/ECD分析,测定结果经常需要进一步用GC/MS或GC/MS/MS做确证分析,如待测物浓度极低时,茶叶色素的背景值会干扰MS全扫描质谱图进行对比分析。而以GC/MS/MS进行分析时,分析物经第一级MS电子轰击后,选择主要母离子或特征离子,以适当碰撞诱导解离(CID)能量做第二次MS电子轰击,产生清晰的MS/MS质谱图,大幅增加信噪比,可做低浓度确证分析。在该方法中,51种分析物可同时进入GC,通过非极性毛细管柱(如HP5-MS柱)分离,再进入MS做定性定量分析,其定量分析结果得到校正线性范围为0.05~5.0ug/ml,检测极限范围为0.001~0.2ug/ml(依据不同分析物而定)。在0.25、0.75及2.5ug/g添加量回收率中,得到平均回收率为70~120%之间。在实际茶叶样品试验中,经GC/ECD检测为阳性的样品,以GC/MS/MS进行确证和定量分析,大部分样品的GC/ECD分析值与GC/MS/MS分析结果一致。但有少部分样品,GC/ECD检测为假阳性。
2、GC/AED技术
虽然GC/MS在农药多残留分析中很受欢迎,但它仍有局限性。当采用选择离子检测(SIM)或串联质谱(MS/MS)时,方法开发较为耗时,且GC中任何保留时间的漂移都要求各个分析物的保留时间窗口相应移动。这些方法只能检测目标化合物表中所列的农药,还有几百种农药及代谢物可能检测不到。原子发射检测器(AED)是近年飞速发展起来的多元素检测器,它是利用等离子体做激发光源,使进入检测器的被测组分原子化,然后原子被激发至激发态,在跃迁至基态时发射出原子光谱。根据这些光谱的波长和强度即可进行定性和定量分析。所以,AED属光度学检测器。由于它是原子(或原子离子)而不是分子激发后发射光,故称为原子发射检测器。AED具有许多独特的性能和应用,如:
1)、AED可以以选择性和通用性两种方式工作:若用杂原子通道,AED可作为选择性检测器,且其选择性较其他气相色谱检测器(如ECD、FPD等)更高,若用碳、氢通道,AED即为通用性检测器,且灵敏度高于FID;
2)、AED对元素周期表中除氦以外的任何一种元素均可检测,属多元素检测器,可用于测定未知化合物的经验式和分子式;对未知物鉴定,AED是MSD、FTIR的有力补充工具;
3)、由于AED选择性强,可降低对复杂混合物高分辨分离的要求,对未完全分离峰也可分别检测;
4)、由于AED的相对响应因子几乎是恒定的,不用标样也可准确定量。
GC/AED的主要应用之一就是测定各种样品中的残留农药、杀虫剂和除草剂等。GC/AED的各元素选择性通道检测不仅能够解决化合物分离问题,而且可以立即判断出各种化学物质中的元素组成,大大简化了分析程序。有文献报道了一种用于筛选567种农药和可疑内分泌破坏物的方法。该筛选方法建立在保留时间锁定(RTL)的气相色谱新技术基础上,采用基于保留时间和元素含量或检测器响应的数据库检索。这一技术能将被分析物的鉴定缩小到仅有几种可能性的范围之内。进一步的确证则采用GC/MS或采用GC/AED计算化合物元素来完成。由于GC/AED的元素响应几乎与分子结构无关,不依赖于化合物的校准可用来定量所发现的所有农药。选择一种已知浓度的农药标准溶液加入到样品溶液中,获得有关元素的特征校正曲线,利用这些曲线来校正任何含一种或多种这些元素的化合物。因为GC/AED相当稳定,外标法可以取得满意的结果。利用这一方法已分析了许多种水果和蔬菜样品,具有多方面的适应性和潜在用途。农药几乎总是含有杂原子,并且一个分子中往往含有几个,最常见的杂原子有O、P、S、N、Cl、Br和F。GC与AED结合已证明是一种非常有用的农药筛选工具,因为它对农药化合物中发现的所有元素都有选择性。遗憾的是,由于种种原因,仪器制造商目前已经停止了这种检测器的生产。但我个人认为,应用这一技术检测农药残留量的方法,给我们带来很多启迪,值得借鉴,其中的一些技术还将做重点介绍。
3、酶抑制和免疫检测技术
从二十世纪八十年代开始,农药的酶抑制和免疫检测技术作为快速筛选检测方法受到许多发达国家的高度重视, 成为农业生物技术领域一个重要分支,得到了快速发展。酶抑制、酶联免疫(ELISA)、放射免疫(RIA)、单克降抗体等技术由于可以避免假阴性,适宜于阳性率较低的大量样品检测,在农药残留检测中应用增多。如依据有机磷和氨基甲酸酯类农药抑制生物体内乙酰胆碱酯酶的活性来检测上述两类农药的残留。
酶抑制和免疫检测技术对所测样品的前处理要求简单,多数样品可直接用于测试。目前,研制成功了多达100多种农用药物检测试剂盒,其中常见的农药残留监测试剂盒有近30种。欧、美、日、巴西、印度等10多个国家,运用这一技术开展了对农产品中有毒物质残留的生物技术监测研究,粗筛检测水产品、肉类产品、果蔬产品中农药残留量。最近,英国研制的通用型有机磷杀虫剂免疫检测药盒可对一些样品中8种以上的有机磷农药进行同时检测。近来我国市场上也广泛推荐和大规模地应用这一快速检测技术。
-
+关注
私聊
-
yhl-87_
第78楼2009/11/06
农药残留检验方法系列讲座(60)(3)农药残留检测与样品前处理技术的发展趋势
4、其他色谱分析新技术
(1)保留时间锁定(RTL)
所谓保留时间锁定,就是使特定化合物的保留时间在不同仪器、不同色谱柱(但标称固定相和相比相同)之间保持不变。决定保留时间的因素主要是化合物的性质、固定液的性质和操作条件。如果前两个因素不变(对于特定的化合物和GC仪器系统,这当然是成立的),那就只有操作条件了,即载气流速、柱温、毛细管柱规格和检测器类型。只要仪器的载气和温度控制精度足够高,只要色谱柱的标称规格一致,就可以通过调节柱前压的方法来补偿操作参数的微小变化,从而实现保留时间的重现,这就是RTL的基础。
现在,随着仪器制造水平和色谱柱制造工艺的进步、仪器自动化程度的提高,各种操作参数的控制更为严密,同一台仪器的保留时间重复性可达到0.01min。但不同仪器、不同色谱柱之间的重现性仍不能令人满意。最近,保留时间锁定(RTL)技术的出现,给这一问题的解决提供了较为理想的途径。保留时间锁定(RTL)技术的应用,使方法中增加更多化合物的检测变得更为简单,只需在相同的锁定条件下确定它们的保留时间即可。带有数据库检索功能的保留时间锁定很容易使该方法应用到相似的分析类型中去。在GC/AED筛选567种农药和可疑内分泌破坏物的方法中,就谈到该方法的一个关键是气相色谱中保留时间锁定技术的研究,采用RTL技术,可以用一个给定的GC方法测定农药的保留时间,然后,在此后的运行中在同一台仪器或不同仪器上重现这些保留时间。由于保留时间的精密度和预测性提高了,使保留时间成为一个极为有用的化合物定性的参数。可以弥补由于不同时间、不同色谱柱和不同仪器差异造成的保留时间的不可预见性。农药多残留检测,要求有一个能使几十种,乃至几百种农药在适当时间流出、同时得到充分分离的GC方法。利用该技术建立一个锁定保留时间表,在不同条件下重现这些保留时间。目前,某些仪器公司已开发出很多的技术软件包括保留时间锁定软件、方法转换软件和农药保留时间表。现在市场上可以买到现成的保留时间锁定软件(RTL),它就是根据上述原理设计和工作的,当一个分析方法确定以后,首先进行一次锁定。用户只要将5个压力下目标化合物的保留时间数据输入计算机,软件就会自动给出p-TR日曲线,并同方法一起储存起来。当在另一台仪器上重复该方法时,只要将原方法拷贝到新仪器上,就可以重新进行锁定。计算机可依据试运行的结果自动计算并调节柱前压,从而使新仪器上的保留时间与方法开发时保留时间很好吻合。虽然目前这种软件只能在HP(现已改为安捷伦)化学工作站中运行,但估计很快会有较为通用的RTL软件出现。
(2)大体积进样技术
目前的进样技术主要有:大口径毛细管柱接口、分流/不分流进样、冷柱上进样和程序升温进样口(PTV),以及基于冷柱上进样和程序升温进样口(PTV)技术的大体积进样和直接进样杆进样。直接进样杆进样适合于高沸点化合物的分析测定,同时直接进样杆结合PTV进样口进行测定,大大减少了样品的前处理工作,适合于对果蔬中农药残留等复杂样品体系的测定,扩大了GC/MS的应用范围,是目前各仪器公司争相开发的一个重点。此外,固相微萃取技术(SPME)也是目前各种GC/MS生产厂家研究的一个重点。由于固相微萃取技术可部分或全部替代顶空进样器、吹扫捕集进样器、液液萃取、液固萃取等技术,且使用更方便,无需溶剂,节省大量的时间和日常操作费用,特别在复杂样品体系的分析中表现出强大的生命力和广阔发展前景,已成为GC/MS的重要配件之一。今天主要为大家介绍基于PTV进样技术的大体积进样。
样品浓缩是提高灵敏度的成熟方法,对许多农药残留分析来说,样品浓缩包括在提取之后的溶剂蒸发,这会产生大量的废溶剂,且大大增加了样品的制备时间。近来科学的发展,比如固相萃取(SPE)、超临界萃取(SFE)、固相微萃取技术等,可避免使用液/液萃取,但是这些方法仍然要增加样品的处理时间。通过降低系统的本底和干扰可以降低检测限,使用高选择性高灵敏度检测器也可以降低检测限。近年发展起来的大体积进样(LVI)技术更是一种有效提高灵敏度的方法。采用比常规GC大几十到几百倍的进样量(5~500 μL)就可提高灵敏度一到两个数量级。实现大体积进样的方式,一是基于冷柱上进样,二是基于PTV技术。文献报道的LVI应用多数是采用程序升温汽化(PTV)进样技术的。这主要是因为PTV进样时样品是在衬管中汽化的,不挥发物滞留在衬管中可以保护色谱柱不被污染,故很适合于分析农药残留量。对于大体积进样,PTV用于“溶剂放空”或“溶剂排除”模式。样品注入进样口,其温度接近溶剂的沸点,且具有相对高的分流比。溶剂(以及低沸点溶质)被放空,而高沸点溶质(约比溶剂沸点高100℃)则保留在进样口中并被浓缩。在到达预先设定的时间后,关闭分流出口,并升高进样口温度以将溶质和残留的溶剂转移到色谱柱进行分离。因为样品是在进样口中汽化,不挥发物和降解产物留在进样口中,这样就最大限度地减少了对色谱柱的污染。研究证明用PTV进样造成的进样口污染对下一次进样的影响要比汽化室加热所造成的影响小。如果进样口被污染,进样口中的衬管很容易更换。所以,对于不干净样品的分析,PTV是比冷柱上进样和分流/不分流进样更好的选择。当然,目标化合物中含有高挥发性组分时,通过PTV做LVI并不是一种好的选择,因为低沸点组分会随同溶剂一起放空而流失。要使分析获得成功,最低沸点目标化合物的沸点应高于溶剂沸点100℃。
三、前处理技术
1、固相萃取(Solid Phase Extraction SPE)
固相萃取(SPE)就是利用固体吸附剂将液体样品中的目标化合物吸附,与样品的基体和干扰化合物分离,然后再用洗脱液洗脱或加热解吸附,达到分离和富集目标化合物的目的。
与液/液萃取相比固相萃取有很多优点:固相萃取不需要大量互不相溶的溶剂,处理过程中不会产生乳化现象,它采用高效、高选择性的吸附剂(固定相),能显著减少溶剂的用量,简化样品的预处理过程,同时所需费用也有所减少。一般说来固相萃取所需时间为液/液萃取的1/2,而费用为液/液萃取的1/5。但其缺点是目标化合物的回收率和精密度要略低于液/液萃取。
固相萃取实质上是一种液相色谱分离,其主要分离模式也与液相色谱相同,可分为正相(吸附剂极性大于洗脱液极性),反相(吸附剂极性小于洗脱液极性),离子交换和吸附。固相萃取所用的吸附剂也与液相色谱常用的固定相相同,只是在粒度上有所区别。
固相萃取选择分离模式和吸附剂时还要考虑以下几点:
1)、目标化合物有极性或非极性溶剂中的溶解度,这主要涉及淋洗液的选择。
2)、目标化合物有无可能离子化(可用调节PH值实现离子化),从而决定是否采用离子交换固相萃取。
3)、目标化合物有无可能与吸附剂形成共价键,如形成共价键,在洗脱时可能会遇到麻烦。
4)、非目标化合物与目标化合物在吸附剂上吸附点的竞争程度,这关系到目标化合物与干扰化合物能否很好分离。
最简单的固相萃取装置就是一根直径为数毫米的小柱,小柱可以是玻璃的,也可以是聚丙烯、聚乙烯、聚四氟乙烯等塑料,还可以是不锈钢制成的。为了方便固相萃取的使用,很多厂家还研制开发了很多固相萃取的专用装置,固相萃取仪。固相萃取的一般操作程序分为如下几步:活化吸附剂:在萃取样品之前要用适当的溶剂淋洗固相萃取小柱,以使吸附剂保持湿润,可以吸附目标化合物或干扰化合物。不同模式固相萃取小柱活化所用溶剂不同:
----反相固相萃取所用的弱极性或非极性吸附剂,通常用水溶性有机溶剂,如甲醇淋洗,然后用水或缓冲溶液淋洗。也可以在用甲醇淋洗之前先用强溶剂(如己烷)淋洗,以消除吸附剂上吸附的杂质及其对目标化合物的干扰。
----正相固相萃取所用的极性吸附剂,通常用目标化合物所在的有机溶剂(样品基体)进行淋洗。
----离子交换固相萃取所用的吸附剂,在用于非极性有机溶剂中的样品时,可用样品溶剂来淋洗;在用于极性溶剂中的样品时,可用水溶性有机溶剂淋洗后,再用适当pH值,并含有一定有机溶剂和盐的水溶液进行淋洗。
为了使固相萃取小柱中的吸附剂在活化后到样品加入前能保持湿润,应在活化处理后在吸附剂上面保持大约1mL活化处理用的溶剂。
-
+关注
私聊
-
yhl-87_
第79楼2009/11/06
农药残留检验方法系列讲座(60)(4)农药残留检测与样品前处理技术的发展趋势
固相萃取技术的应用:固相萃取主要用于复杂样品中微量或痕量目标化合物的分离和富集。例如,生物体(如血液、尿等)中药物及其代谢产物的分析,食品中有效成分或有害成分的分析,环保水样中各种污染物(可挥发性有机物和半挥发性有机物)的分析都可使用固相萃取将目标化合物分离出来,并加以富集。
2、固相微萃取(SPME)
固相微萃取(SPME)是在固相萃取上发展起来的一种新型、高效的样品预处理技术,它集采集、浓缩于一体,简单、方便、无溶剂,不会造成二次污染,是一种有利于环保的很有应用前景的预处理方法。与液/液萃取和固相萃取相比,具有操作时间短,样品量小,无需萃取溶剂,适用于分析挥发性与非挥发性物质,重现性好等优点。很多研究结果表明,在样品中加入适当的内标进行定量分析时,其重现性和精密度都非常好。固相微萃取装置外形如一只微量进样器,由手柄(holder)和萃取头或纤维头(fiber)两部分构成,萃取头是一根1cm长,涂有不同吸附剂的熔融纤维,接在不锈钢丝上,外套细不锈钢管(保护石英纤维不被折断),纤维头在钢管内可伸缩或进出,细不锈钢管可穿透橡胶或塑料垫片进行取样或进样。手柄用于安装或固定萃取头,可永远使用。固相微萃取的萃取过程是一个平衡过程,萃取的平衡时间与搅拌速度、固定相的膜厚以及被分析样品的分配常数、扩散系数、萃取温度有关。大分子质量的物质比小分子质量的物质需更长的分析时间。搅拌有利于减少达到平衡所需时间,当达到平衡时,固相微萃取方法的灵敏度最高。
固相微萃取技术包括两个过程:(1)样品中待分析物在石英纤维上的涂层与样品间扩散、吸附、浓缩过程以及浓缩的待分析物脱附进入分析仪器完成分析过程。在前一个过程中,涂有吸附剂的石英纤维浸入样品中,使样品中目标化合物从样品基质可中扩散、萃取、浓缩于涂层上。(2)将石英丝收回针头中,进样时直接插入分析仪器的进样室中,如气相色谱仪的汽化室,使萃取的化合物脱附,被载气导入色谱柱完成分离分析。在实施固相微萃取时可以采用直接固相微萃取法、液上空间固相微萃取法和衍生化固相微萃取法3种模式。影响固相微萃取灵敏度的因素很多,但萃取头涂层种类和厚度对灵敏度的影响最为关键。一般来说,不同种类待测物要用不同类型的吸附质涂层进行萃取,其选择基本原则是“相似相溶原理”。用极性涂层萃取极性化合物,用非极性涂层萃取非极性化合物。
3、微波萃取技术(MAE)
微波萃取就是利用极性分子可迅速吸收微波能量来加热一些具有极性的溶剂,如乙醇、甲醇、丙酮和水等。因非极性溶剂不能吸收微波能量,所以在微波萃取中不能使用100%的非极性溶剂作为萃取溶剂。一般可在非极性溶剂中加入一定比例的极性溶剂来使用。如丙酮-环己烷(体积比=1:1)就可用来做微波萃取溶剂。微波萃取是将样品放在聚四氟乙烯材料制成的样品杯中,加入萃取溶剂后将样品杯放入密封好、耐高压又不吸收微波能量的萃取罐中。由于萃取罐是密封的,当萃取溶剂加热时,由于萃取溶剂的挥发使罐内压力增加。压力的增加使得萃取溶剂的沸点也大大增加,这样就提高了萃取温度。同时,由于密封,萃取溶剂也不会损失,也就减少了萃取溶剂的用量。
微波萃取装置一般是一台带有控温和控时的微波加热装置,根据需要选用体积为50~100ml的聚四氟乙烯材料制成的样品杯和放样品杯的密封罐。影响微波萃取效果的主要因素有萃取温度、萃取溶剂、样品杯材料吸附及记忆效应等。
4、加速溶剂萃取(ASE)
加速溶剂萃取(ASE)是一种全新的处理固体和半固体样品的方法,该法是在较高温度(50~200℃)和压力条件(10.3~20.6MPa)下,用有机溶剂萃取。它的突出优点是有机溶剂用量少(1g样品仅需1.5mL溶剂)、快速(一般为15min)和回收率高,已成为样品前处理最佳方式之一,并被美国EPA(环保局)选定为推荐的标准方法(标准方法编号EPA3545),已广泛用于环境、药物、食品和高聚物等样品的前处理,特别是农药残留量的分析。
在提高的温度下能加速溶质分子的解析动力学过程,减小解析过程所需的活化能,降低溶剂的粘度,因而减小溶剂进入样品基体的阻力,增加溶剂进入样品基体的扩散。已报道温度从25℃增至150℃,其扩散系数大约增加2~10倍,降低溶剂和样品基体之间的表面张力,溶剂能更好地“浸润”样品基体,有利于被测物与溶剂的接触。液体的沸点一般随压力的升高而提高。例如,丙酮在常压下的沸点为56.3℃,而在0.5MPa时,其沸点高于100℃。液体对溶质的溶解能力远大于气体对溶质的溶解能力。由于加速溶剂萃取是在高温下进行,因此,热降解是一个令人关注的问题。加速溶剂萃取的运行程序是先加入溶剂,即样品在溶剂包围之下,再加温,而且在加温的同时加压,即是在高压下加热,高温的时间一般少于10min,因此,热降解不甚明显。
加速溶剂萃取仪由溶剂瓶、泵、气路、加温炉、不锈钢萃取池和收集瓶等构成,其工作程序的第一步是手工将样品装入萃取池,放在圆盘式传送装置上,以下步骤按先后全自动进行,即圆盘传送装置将萃取池送入加热炉腔并与对应编号的收集瓶联接,输液泵将溶剂输送到萃取池(20~60s),萃取池在加热炉中被加温和加压(5~8min),在设定的温度和压力下静态萃取5min,分别少量向萃取池加入清洗溶剂(2~60s),萃取液自动经过滤膜进入收集瓶,用N2吹洗萃取池和管道(60~100s),萃取液全部进入收集瓶,待分析。全过程仅需13~17min。溶剂瓶由4个组成,每个瓶可分别装入不同的溶剂,可选用不同溶剂先后萃取相同的样品,也可用同一溶剂萃取不同的样品。可同时装入24个萃取池和26个收集瓶。ASE200型萃取仪,其萃取池的体积可从11mL~33mL。ASE300型萃取仪的萃取池体积可选用33mL、66mL和100mL。
5、凝胶渗透色谱(GPC)
凝胶渗透色谱技术被用来去除脂肪和其它分子量相对较高的化合物,适用的样品范围极广,回收的农药品种多,回收率也较高,不仅对油脂净化效果好,而且分析的重现性好,柱子可以重复使用,已成为农药多残留分析中的通用净化方法。其作用类似一组分子筛,将样品溶液加到柱子上后,用溶剂淋洗,分子量大于农药的类脂肪、色素、蜡质等先被淋洗出来,然后农药按分子量大小相继被淋洗出来。常用的淋洗剂有环己烷/二氯甲烷、甲苯/乙酸乙酯、二氯甲烷/丙酮等。凝胶渗透色谱技术在欧美国家应用非常普遍。
采用多残留检测技术和快速筛选检测技术,结合各种先进的、各具特色的前处理技术检测食品中农药残留量已成为当今的发展趋势。迄今为止,许多传统的样品前处理方法和技术得到了进一步改进,新的处理方法和技术也相继出现,快速、有效、简单、绿色的色谱分析样品处理方法和技术成为色谱工作者的追求。
(全文完)
另外: 该文《农药残留检测与样品前处理技术的发展趋势》介绍的方法比较全面,因而吸收过来将jpg格式转化为doc,系本网的文章待查出处。
-
+关注
私聊
-
yhl-87_
第80楼2009/11/06
农药残留检验方法系列讲座(61)固相萃取-高效液相色谱法测定苹果中
残留的克菌丹和灭菌丹
摘 要:采用硅镁吸附剂和硅胶作吸附剂,建立了固相萃取一高效液相色谱法同时测定苹果中残留的克菌丹和灭菌丹的分析方法。研究了甲醇一乙腈一水(含0.1 mmol/L乙酸-乙酸钠缓冲溶液pH 3.80)三元体系下的克菌丹和灭菌丹的最佳分离条件,在波长210 nm下检测,克菌丹和灭菌丹的线性范围为0.40~8.00mg/kg,线性相关系数均大于0.9999;最低检出限克菌丹为0 .27 mg/kg、灭菌丹为0.20 mg/kg;保留时间的相对标准偏差(RSD)≤0.60%) 。苹果样品中3个添加水平的平均加标回收率为克菌丹69.3%~106%,RSD为3.7%~4.7%;灭菌丹101%~l08%,RSD为1.3%~5.4%。
前 言
克菌丹(captan)和灭菌丹(folpet)属于三氯甲硫基类杀菌剂,是苯二甲酰亚胺的衍生物。克菌丹和灭菌丹主要用于杀菌,它们对多种果树、蔬菜病害的防治效果好,药害小,适当使用还有刺激植物生长的作用。我国无公害食品苹果中克菌丹的限量标准为低于5 mg/kg 欧盟食品农药残留限量标准规定苹果中克菌丹和灭菌丹的含量要低于3 mg/kg,其他蔬菜如黄瓜、茄子要低于0.l mg/kg
测定克菌丹和灭菌丹的方法主要有薄层色谱法[1] 高效液相色谱法(H PLC〉[2]、气相色谱法[3,4] 和气相色谱-质谱联用法[5] 。日前国内测定水果中克菌丹和灭菌丹的标准方法分别为中华人民共和国进出口商品检验行业标准SN0654-1997[6]和SN0191-93[7]。SN0654-1997方法是用高效液相色谱法测定水果中的克菌丹,方法的加标回收率很低,灵敏度无法满足克菌丹残留限量分析的要求。文献[4] 及SNOl9l-93的方法均是用气相色谱法分别测定克菌丹和灭菌丹,两种方法的提取步骤比较繁琐。我国目前尚未建立高效液相色谱法同时测定水果中克菌丹和灭菌丹的检验标准 为此本文建立了采用高效液相色谱同时测定苹果中克菌丹和灭菌丹的分析方法,研究了用国产硅镁吸附剂和硅胶混合吸附剂固相萃取净化样品的条件。
本文由王淑菊l, 于彦彬2, 谭培功3, 苗在京2, 魏亦山2 (1中国海洋大学;2农业部农产品质量安全监督检验测试中心;3青岛市环境监测中心站)共同完成。他们通过实验对SN0654-1997[6]和SN0191-93[7]。SN0654-1997方法提出了一些修改意见。现全文介绍如下供参考:
实 验
1 实验部分
1.1 仪器与试剂
Waters Alliance 高效液相色谱系统,带有2487双波长紫外检测器(Waters公司);超纯水系统(北京历元电子仪器公司);IKAR ULTRA TURRAX T18 basic分散机(广州仪科实验室技术有限公司);Cary-100 Conc紫外-可见分光光度计(Varian公司)。l 000 mL流动相过滤装置,0.45 μm水相、有机相滤膜。
克菌丹标准物为固体,纯度为99.4%(Dr. Ehrenstorfer公司,德国),准确称取50.24 mg克菌丹标准物置50 mL烧杯中,用甲醇溶解并转移到50mL容量瓶中,用甲醇稀释至刻度,得到质量浓度为1.00 g/L的标准液。灭菌丹标准物为固体,纯度为99.8%(SIGMA-ALDRICH LABORCHEMIKALIENGMBH)。准确称取50.l0 mg灭菌丹标准物质置50mL烧杯中,用甲醇溶解并转移到50 mL容量瓶中并稀释至刻度,得到质量浓度为1.00 g/L的标准液。便用时用甲醇将克菌丹和灭菌丹标准液稀释为合适浓度的混合溶液c
正已烷、丙酮、二氯甲烷、环己烷、乙酸乙酯为分析纯,使用前用全玻璃蒸馏器重蒸。乙腈为色谱纯。
乙酸-乙酸钠缓冲溶液(pH 3.25):浓度为0.01mol/L,使用时纯水稀释100倍,以0.45μm水相滤膜过滤。
无水硫酸钠(分析纯):在500℃马弗炉中灼烧4 h,备用。
氯化钠(分析纯):在140℃烘箱中烘4 h。
硅镁吸附剂(粒度为 75~150 μm,中国医药集团上海化学试剂公司):在650℃马弗炉中灼烧3h,然后在I30℃烘箱中烘3 h,再加人其质量的l0%的水去活,平衡24 h后可使用。
硅胶(粒度为75~150μm,青岛胜海化工有限公司):在130℃烘箱中烘3 h后,加入其质量的l0%的水去活,平衡24 h后可使用。
1.2 色谱条件
色谱柱:ZORBAX Eclipse XDB-C18 柱(150 mm×4.6 mm×5μm),Waters Symmetry ShieldTM RP18柱(150 mm×3.9 mm×5μm)。流动相:甲醇-乙腈-水(含0.1 mrnol/L乙酸-乙酸钠缓冲溶液(pH3.80)),流速1.0 mL/min。柱温为30℃,检测波长为210 nm,进样量为20μL。
1.3 实验方法
1.3.1 净化柱的制备
在l0 mL的玻璃注射器内放人少许脱脂棉,从下到上依次分别加人0.15 g硅镁吸附剂、0.75 g硅胶和0.5 g左右的无水硫酸钠,敲击注射器使其装填均匀。加5.0 mL丙酮- 正己烷(体积比为1:9)混合溶液于玻璃注射器中,润湿吸附剂以备用于样品净化。
1.3.2 样品的处理
将25.00 g加工好的匀浆样品放人100 rnL烧杯中,加水15.0 mL、磷酸-水(体积比为1:1)溶液2.0 mL、乙腈50.0 mL,在分散机中匀浆2 min,然后过滤到装有5~7 g氯化钠的100 mL具塞量筒中,收集滤液60~70 mL,盖上塞子,剧烈振荡1min,在室温下静置20min,待乙腈相和水相分层。准确吸取l0.00 mL乙腈提取液,置于50 mL烧杯中,将烧杯放在70℃的水浴上加热、蒸发至近干。在烧杯中加人2.0 mL正己烷溶解样品,然后将溶液转移到净化柱中净化,用5.0 mL正己烷-二氯甲烷-乙腈(体积比为50:4⒐1)混合溶液洗烧杯,并加到柱中洗脱,再重复两次,收集所有洗脱液。将洗脱液在旋转蒸发仪上浓缩至约5 mL,浓缩温度为72℃,然后用氮气流吹干,再用甲醇溶解并定容至2.00 mL,供HPLC分析。
2 结果与讨论
2.1 色谱柱的选择
试验表明选用Waters Symmetry ShieldTM RP18色谱柱和ZORBAX Eclipse XDB-C18色谱柱均能分离克菌丹和灭菌丹,但Eclipse XDB-C18色谱柱的分离度优于Symmetry Shield TM RP18柱,因此选用Eclipse XDB-C18柱作分析柱。
2.2 流动相的优化
在优化流动相甲醇一乙腈-水三元体系时发现,在中性条件下,灭菌丹有两个色谱峰,第一个色谱峰的保留时间与克菌丹相差不足0 1 min,信号大小与克菌丹也相近;当调节水相的pH值为3.0时,灭菌丹的第一个色谱峰的响应值大大减小。这可能是因为灭菌丹在中性条件下分解造成的。Martinez等[8]用乙腈-水(体积比为45:55)作流动相,电化学检测器分析克菌丹和灭菌丹时,水相含有0.01mol/L的乙酸-乙酸钠缓冲溶液。本文试验发现用0.01 mol/L的乙酸一乙酸钠缓冲溶液作流动相,以紫外检测器检测,空白值高,改用0.1 mmol/L的乙酸一乙酸钠作缓冲溶液时,空白值可降至不影响测定的灵敏度,因此确定三元流动相屮各相的体积分数为甲醇50%、乙腈5%、水45%(含有0.l mmol/L的乙酸-乙酸钠缓冲溶液)。在此条件下等度分离,标准溶液和实际样品色谱图见图1。由图1可见,克菌丹和灭菌丹的两个峰均能完全达到基线分离。