-
+关注
私聊
-

yhl-87_
第11楼2009/11/06
薄层色谱法应用系列讲座(11)
薄层色谱展开体系的筛选
一、 展开剂、吸附剂和被测成分的关系
薄层色谱中的流动相即为展开剂,其溶质即为被测成分,吸附剂即为薄层板上涂敷的固定相所以薄层色谱基于液-固吸附色谱,其溶质的保留和分离选择性决定于三个因素,如下图
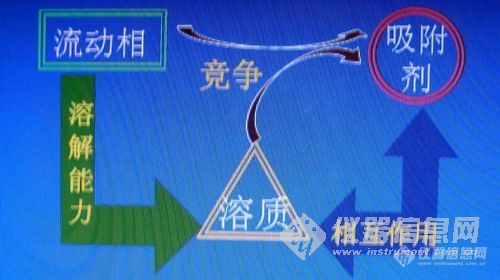
实际上它们三者相互竞争、相互致约在不断变化的复杂体系中
* 液-液分配
* 基于组分在互不相溶的两相间的溶解度不同而实现分离的一种展开系统。可在展开前,将薄层板浸入某种液体中。实际上在TLC中,分配与吸附是并存的(大气中水分也是一种影响因素)。
* 还有化学键合相反相展开、化学键合相正相展开、离子交换、离子对色谱、凝胶、胶束、手性展开等
二、溶剂强度参数和选择性
⒈ 溶剂的强度参数用°ε表示
溶剂强度参数°ε越高,表示它与吸附剂相互作用力强。溶质的Rf值就越大。为得到满意的分离,选用的流动相使溶质的Rf值在0.3 – 0.7之间为宜。
溶剂强度也可用其在水中的溶解度参数d 来表示,参考下面部分溶剂参数值表。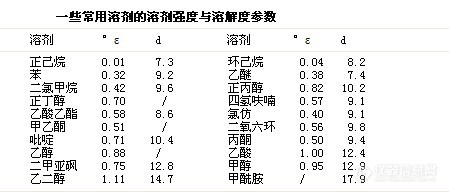
三、 展开剂选择时需考虑的问题
a) 一些溶剂如氯仿、乙醚一般含有微量乙醇作保护剂,有时需重蒸除去乙醇。
b) 许多溶剂有吸湿性,使溶剂含水量不一致,导致实验重现性差。应进行除湿干燥处理。如用无水硫酸钠过滤等。
c) 溶剂储藏时间和条件对溶剂质量影响,应注意出厂日期。
d) 混合展开剂组分之间(或杂质)可能发生相互作用。
e) 对于易挥发的溶剂,难于配制稳定组成的展开剂。要求操作格外小心,使用过的混合流动相不应重复使用,因用过的展开剂其各组分比例已有变化。
f) 分离度指溶剂能引起两种溶质位移差别的能力。
R=1/4(a -1)[`k /(k +1)]
分离因子a= k1/k2,
k为k1和k2的平均值
由上式可见,R取决于三个因子:
① 理论塔片n主要是吸附剂性质的函数
② `k 反映了两种溶质的平均迁移速度,其由流动相溶剂强度°ε 决定
③ a 取决于吸附剂和流动相的组成
四、 参考溶剂优化的某些规则
a) 增加溶剂强度,使Rf值增大,但也可能降低分离能力。
b) 流动相中较强组分的体积比小于5%或大于50%,选择性最大,此时最有利于溶质与流动相组分的选择性相互作用。
c) 用乙醚或甲醇代替流动相中的一种较强组分,由于形成氢键可改善选择性。
d) 若出现斑点拖尾,可向流动相中加少量水,或降低薄层活性,有利于改善分离。
e) 也可采用两种强溶剂与一种弱溶剂组成的三元混合流动相,此时,溶剂强度由弱溶剂控制,而溶剂选择性则由两强溶剂控制。
-
+关注
私聊
-
yhl-87_
第12楼2009/11/06
薄层色谱法应用系列讲座(12)
高效薄层扫描法测定
蛛网膜下腔出血患者脑脊液中血小板活化因子
’
摘要 建立了测定蛛网膜下腔出血患者(SAH)脑脊液中血小板活化因子(PAF)质量浓度的高效薄层扫描方法。薄层板为高效硅胶G板(100mm×100mm),展开剂为V(氯仿):V(甲醇):V(水)= 65:35:6,下层液展开,显色剂为100g/L磷钼酸溶液,展开距90mm,扫描波长630nm。方法的线性范围0.5~2.5μg/L,相关系数0.9990,最小检测质量浓度50ng/L,平均回收率98.6%。运用所建立的方法测定了16例SAH患者发病后脑脊液和l0例非神经系统疾病患者脑脊液中PAF的质量浓度及其变化规律,为临床提供了诊断依据。
1 前言
血小板活化因子(platelet:activating factor,PAF)是一种脂类介质。它是1972年由Benveniste等⑴ 研究白兔过敏反应过程中发现的。后来Lindsberg等⑵ 研究兔的脊髓缺血组织发现,缺血神经组织的PAF水平比正常的高20倍,中枢神经组织尤其是缺血的神经组织亦可产生大量的PAF。因此PAF与脑缺血之间的关系越来越受到重视⑶-⑹。但测定脑脊液中PAP、的方法未见文献报道。本文建立了一种特异、灵敏、准确测定蛛网膜下腔出血(SAH)患者脑脊液中PAF质量浓度的方法,有助于阐明脑缺血的某些病理机制,为其防治提供了新的依据,测定的结果满意。本文是由兰州军区后勤部药物研究所的王荣、赵兴红、兰州军区总医院的张新江和兰州军区医学高等专科学校的胡晓丽共同完成。现全文介绍如下。
2 材料和方法
2.1 仪器及试药
岛津CS-920型薄层扫描仪;岛津U-135C型积分仪。
PAF标准对照品,购自美国Sigma公司;高效硅胶G板,青岛海洋化工厂;其余试剂均为分析纯。
2.2 色谱条件
高效硅胶G板;展开剂为V(氯仿):V(甲醇):V(水)= 65:35:6,摇匀放置过夜,用下层液展开;显色剂:100g/L磷钼酸溶液;展开距10cm;扫描波长630nm;点样量3μL。
2.3 样品处理
蛛网膜下腔出血组:16例(男10例,女6例)蛛网膜下腔出血(SAH)发病患者(诊断符合1986年全国脑血管会议标准,均经头颅CT扫描证实),发病后1~3、7~10、14~21天腰穿采集脑脊液3mL,将采集的脑脊液立即离心,取上清液1mL。加入甲醇5mL振摇,再次离心。离心后加入氯仿4mL和水3mL充分振摇,离心分出氯仿相,-20℃下保存。对照组:10例(男7例,女3例)为同期住院手术的非神经系统疾病患者,无心、脑、肾、肺等疾病。腰麻时收集脑脊液,脑脊液处理的方法同上。薄层展开时,将真空抽干保存于-20℃下的样品标本用氯仿溶解,容量瓶定容。
3 结果
3.1 展开
在上述色谱条件下点样3μL,薄层展开图及薄层扫描图见图-1。
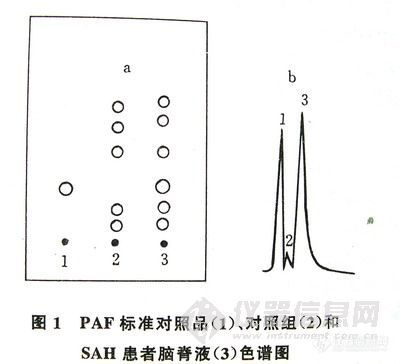
图-1 PAF标准对照品(1)、对照组(2)和SAH患者脑膏液(3)色谱图
a-脑脊液薄层展开图,b-脑脊液薄层扫描图。
图-1中1,2,3依次为PAF标准对照品、对照组、蛛网膜下腔出血(SAH)发病患者脑脊液。展开行为良好,斑点边缘清晰,无拖尾现象。
3.2 标准曲线
在同一块高效板上用PAF、标准对照品点样6个点,使其质量浓度为0.5,1.0,1.5,2.0,2.5,3.0μg/L。经展开后显色、扫描,测定斑点峰面积,求得回归方程为Y=0.294X+406.000,Y为峰面积,X为点样的质量浓度,相关系数为r =0.9990,测得的最小质量浓度为50ng/L。3.3 方法精密度及回收率
在同一块高效板上,点一定质量浓度的PAF标准对照品,共6个点,按上述方法展开、显色、扫描。每隔0.5h依法在不同板上点样,测定日内结果见表-1。同时连续5天依法在不同板上点样,测定日间误差见表-1。回收率是在同一块高效板上分别点上一定量的发病组脑脊液、发病组脑脊液加标准对照品、标准对照品(点两个剂量的不同点)共4个点,按上述方法展开、显色、扫描,计算PAF的量,测得回收率为98.6% (n =5),结果见表-1。该方法精密度好,日内误差为1.96%,日间误差为2.06%。
表-1测定方法的精密度和回收率(n=5)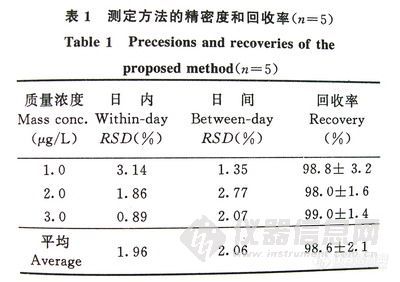
3.4 稳定性试验
PAF稳定性试验包括两部分,一部分是PAF从脑脊液中提取后在冰箱中-20℃冷冻保存不同时间的稳定性,另一部分是PAF配制成氯仿溶液后在室温下放置不同时间的稳定性。PAF提取后用氮气吹干,样品置于-20℃冰箱中存放,分别于1、5、10、30天取出按试验方法测定3份,RSD为3.8%,表明PAF样品在-20℃下至少可保存一个月。将上述保存于-20℃冰箱中的脑脊液配置成氯仿溶液,容量瓶密封放置于室温,分别于立即、2天、5天点样,RSD为1.8%,说明配制成氯仿的PAF溶液密封存放于室温下,5天内仍很稳定。3.5 样品脑脊液中PAF的测定
按上述方法测定了16例蛛网膜下腔出血患者的脑脊液及10例对照组脑脊液中的PAF质量浓度,见表-2。16例蛛网膜下腔出血患者的脑脊液中PAF变化规律见图-2。
表-2蛛网膜出血患者及对照组脑脊液中PAF定量分析结果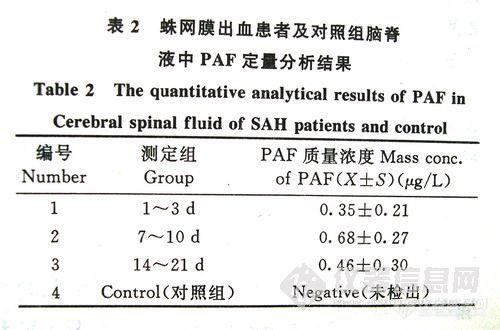
图-2 16例SAH患者脑脊液中PAF质量浓度的变化
4 讨论
PAF半衰期短(仅为3Os),不宜捕获,且正常人的脑脊液中不存在PAF。本文采用高效薄层色谱法,抽取脑脊液后立即用甲醇除去蛋白,破坏乙酰水解酶的活性,直接测定SAH患者发病后脑脊液中的PAF,防止PAF失活,提高了灵敏度。所建立的方法可一次测定多个脑脊液样品,回收率及灵敏度均较高,重复性也好,比HPLC或MS法价廉。同时发现,PAF对酸、温度较稳定,多次用磷钼酸显色,加热至80℃并不影响PAF的活性,与Pinckard等⑺ 报道的结果相符。PAF是目前发现的最强的促血小板聚集物。SAH后极易并发脑血管痉挛,引起脑缺血。研究表明,PAF可引起血脑屏障破坏,脑血管收缩,此外,可通过促使颗粒白细胞释放白三烯等活性物质间接引起脑血管痉挛,使脑血流量减少,故而推测PAF与SAH后脑血管痉挛有关。通过对16例SAH患者发病后1~3天、7~10天、14~21天脑脊液中的PAF测定并与10例非神经系统疾病患者进行了比较发现,对照组脑脊液中PAF均为阴性,由于PAF不能通过血脑屏障,所以正常人脑脊液中不含PAF及其乙酰水解酶。而发病组在不同时期内的PAF质量浓度呈现不同的趋势。初步证明,SAH后脑脊液中PAF的质量浓度明显增加,发病后第二周最明显,说明PAF可能参与了脑血管痉挛、脑缺血的发病过程。也可预测SAH患者易在第二周内发病,与临床实际脑血管痉挛发病在SAH的第二三周高峰时间相吻合。建议临床在第二三周予以防治。本文建立的测定脑脊液中PAF的质量浓度的方法具有一定的临床实际指导意义。
参考文献
1、 Benveniste J,Henson P M,Cohhrane C C T.J Exp Med,1972,138:1356-1357
2、 Lindsberg P J,Yue T L,Frericks K U et a1.Stroke,1990,21:1452—1454
3、 Nishida K,Markey S P.Stroke ,1996,27:514—519
4、 Perhn J H,Krieglstein T.J Neurosei Res,1993,34:179—188
5、 徐运,蒋定国.临床神经病学杂志,1995,8:239~240
6、 Hirashima Y,Endo S,0hmori T.J Neurosurg,1994,80:31-36
7、 Pinckard R N.J Immunology,1979,124(4):1847—1848
-
+关注
私聊
-
yhl-87_
第13楼2009/11/06
薄层色谱法应用系列讲座(13)
薄层色谱法分离测定
1.5-苯并硫氮杂卓-α-氯代-β-内酰胺
,
摘要:用薄层色谱法分离测定反应混合物中的1.5-苯并硫氮杂卓-α-氯代-β-内酰胺。采用透射法锯齿扫描定量分析, 其变异系数小于5%,最低检测限为0.114μg,定量校正线性相关系数0.9952,方法的平均回收率为96.75%,方法简便、快速。
本文是河北省教委资助项目,由河北师范大学化学系的孙娜、李媛、王继业和河北邯郸高等农业专科学校基础部的杜彩云老师们共同完成的,具体介绍如下:
1 前言 苯并硫氮杂卓是一类具有重要生理活性的七员杂环化合物,近年来通过其分子中亚胺双键的环加成反应向分子中引入一个β-内酰胺环,引起人们广泛关注⑴~⑷。已发表的文章中所报道的产率一般都是离析产率,关于产物的定量分析方法尚无报道。本文在研究1.5-苯并硫氮杂卓-α-氯代-β-内酰胺 (简称β-内酰胺) 的合成、立体结构的同时,建立了薄层色谱定量分析方法。该方法简便、快速,且对于各类杂卓β-内酰胺衍生物的定量分析都有一定的参考意义,具有普遍性。
2 实验部分
2.1仪器与试剂
CS-930薄层色谱扫描仪 (日本Shimadzu);λ-254 nm紫外灯(日本Shimadzu);939薄层制板器 (重庆南岸贝尔德仪器技术厂);定量毛细管 (美国Drummond);硅胶60GF254 (青岛海洋化工集团公司);本文所用其它试剂均为分析纯。
2.2 薄层板的制备
取一定量的硅胶60GF254,加入3倍的5g/L羧甲基纤维素钠的水溶液,搅匀后用薄层制板器均匀地铺在20cm×20cm的玻璃板上,厚_0.3mm,自然晾干后置于105℃烘箱中活化1h,然后放人干燥器中备用。
2.3 实验方法
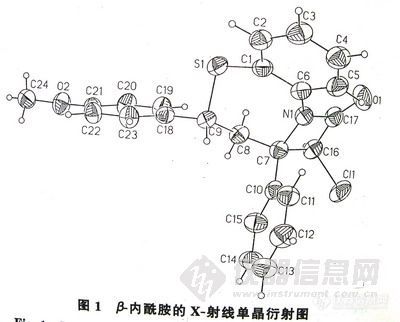
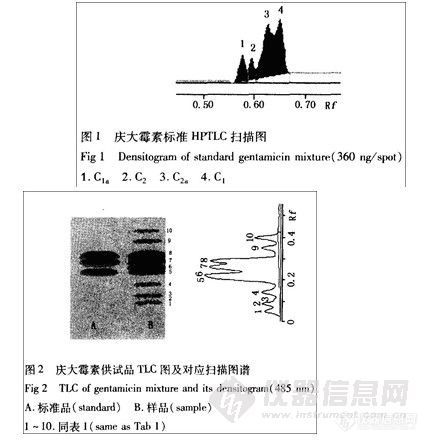
将反应混合物溶于二氯甲烷,用V(乙酸乙酯):V(石油醚) =1:8在室温下上行展开12cm,在λ-254nm紫外灯下照射,显示有6个斑点。经X-射线单晶衍射证实第3个点 (Rf =0.25)是所合成的β-内酰胺 (见图-1)。采用透射法锯齿扫描测量和外标两点法定量,扫描参数为:λ1=270nm,λ2=310nm,狭缝1.2mm×1.2mm,线性仪SX=3。
3 结果与讨论
3.1 β-内酰胺样品的薄层分离
薄层展开后用透射法锯齿扫描,扫描图见图-2。
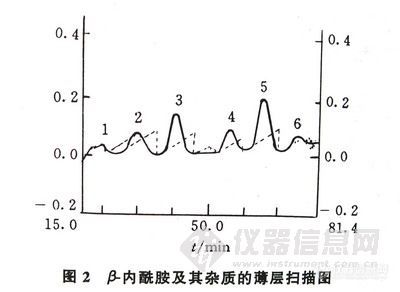
图-2β-内酰胺及其杂质的薄层扫描图
Fig-2 Thin-layer chromatogram of β-lactam and impurities
1、杂蕈 2、开环产物 3、β-内酰胺 4~6、其它杂质。
3.2 工作曲线及检出限
称取自制的β-内酰胺标样5.7mg,用二氯甲烷溶解定容于10mL容量瓶中,配成0.57g/L的标准溶液。点样展开测定,以点样量对峰面积作图,得标准工作曲线,其回归线性方程为Y=1138.92 X +17661.0,r =0.9952 (n=8);线性范围为0.285~5.7μg;最低检出限为0.114μg。
3.3 精密度试验
在同一薄层板上点相同量的斑点,展开测定,其RSD=4.84% (n=6),不同板上相同量的斑点展开测定,其RSD =5.61% (n= 4)。
3.4 样品分析
称取所合成的β-内酰胺样品14.1mg,用二氯甲烷溶解并定容于10mL容量瓶中。在同一薄板上,点0.5、1、2μL等3个标准溶液点和样品溶液点(6×2μL)。平行测定4次,由平均值在标准曲线上求出β-内酰胺,结果见表-1。
表-1样品分析结果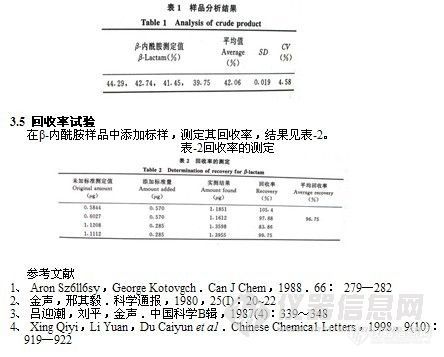
-
+关注
私聊
-
yhl-87_
第14楼2009/11/06
薄层色谱法应用系列讲座(14)
分析小儿清热速崩片质量标准
摘要 目的:建立小儿清热速崩片的质量标准。
方法:采用TLC法对处方中的甘草、黄芩进行定性鉴别,甘草TLC法鉴别以乙酸乙酯一甲酸一乙酸一水(15:1:1:1)为展开利;黄芩TLC法鉴别以醋酸乙酯一丁酮一甲酸-水(5:3:1:1)为展开剂:用HPLC法定量分析麻黄碱的含量,色谱柱为C18(4.6 mm×150 mm×5 μm),以0.l%磷酸溶液(100 mL中加三乙胺0.l mL)一乙腈(99:3)为流动相,检测波长205 nm,流速0.8 mL·min-1。
结果:用TLC法能检出甘草、黄芩:HPLC法测定盐酸麻黄碱在0.06~2.8 μg的范围内呈良好的线性关系,线性回归方程为Y=2.99×103X-15. 9 ,r=0.9999:平均回收率为99.8%,RSD=0.56%。
结论:方法简便、准确,重复性好,能有效控制该制剂质量。
本文由浙江康恩贝制药股份有限公司的郑琚、王紫秋和孙兰香研究人员共同完成现全文介绍如下:
前言
小儿清热速崩片是以麻黄、苦杏仁、石膏、甘草、黄芩、板蓝根、北豆根7味中药制成的复方剂型,具有清热、宣肺、平喘、利咽功效。为了有效控制产品质量,本文建立了TLC法对甘草、黄芩进行定性鉴别,HPLC法对麻黄中麻黄碱进行含量测定。
实验
1 仪器与试药
Agilent 1100高效液相色谱仪;硅胶G板(自制,10 cm×10 cm),对照品盐酸麻黄碱、黄芩苷及对照药材甘草(中国药品生物制品检定所提供),甘草酸铵对照品(康恩贝研发中心提供,经浙江省药品检验所标定,纯度为98.5%),小儿清热速崩片(规格:每片1200 mg康恩贝新研发产品),乙腈为色谱纯,其他试剂均为分析纯。
2 甘草的TLC法鉴别
2.1 对照药材溶液的制备
取对照药材甘草0.5g,加无水乙醚30 mL,加热回流1 h,滤过,弃去乙醚液,残渣挥干溶剂,加甲醇20 mL,加热回流1 h,滤过,滤液蒸干,残渣加水30 mL使溶解。用正丁醇提取3次,每次20 mL,合并提取液,用20mL水洗涤1次,弃去水层,蒸干提取液,残渣加甲醇10 mL使溶解,得对照药材溶液。
2.2 对照品溶液的制备
称取甘草酸铵对照品适量,加甲醇制成每1 ml,含2 mg的溶液,作为对照品溶液。
2.3 供试品溶液的制备
取本品20片研细,称取1g,加水20mL溶解,离心(2000 r·min-l,10min),取上清液移至分液漏斗中,用正丁醇提取3次,每次20mL,合并提取液,用20 mL水洗涤1次,弃去洗液,蒸干,残渣加甲醇l0 mL使溶解,得供试品溶液。
2.4 阴性供试品溶液的制备
照小儿清热速崩片处方制得缺甘草的阴性样品,同供试品溶液制备得阴性供试品溶液。
2.5 薄层色谱法
吸取上述4种溶液各5μL,分别点样于同一块硅胶G薄层板上,以乙酸乙酯一甲酸一乙酸一水(15:1:1:1)为展开剂,展开至8 cm,取出,晾干,喷以l0%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外灯(365 nm)下检视。样品色谱中在与对照品、对照药材色谱相应的位置上分别显相同颜色的斑点,而阴性样品在相应的位置上无斑点。结果见图l。
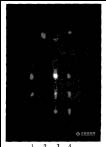
图-1 甘草在荧光下薄层色谱图
1. 甘草酸铵对照品 2. 阴性样品 3. 甘草对照药材 4. 样品
3 黄芩的TLC法鉴别
3.1 对照品溶液的制备
取黄芩苷对照品适量,加甲醇制成每l mL中含1 mg的溶液,作为对照品溶液。
3.2 供试品溶液的制备
取本品20片研细,称取约0.8 g,加甲醇l0 mL,超声处理20 min,离心(2000 r·min-1,15 min),上清液作为供试品溶液。
3.3 阴性供试品溶液制备
照小儿清热速崩片处方取得缺黄芩的阴性样品,同供试品溶液制备得阴性供试品溶液。
3.4 薄层色谱法
吸取上述3种溶液各10 μL,分别点样于同一块硅胶G薄层板上,以醋酸乙酯一丁酮一甲酸一水(5:3:1:1)为展开剂,展开至8 cm,取出,晾干,喷以I%三氯化铁乙醇溶液。样品色谱图中在与对照品色谱相应的位置上显相同的暗绿色斑点,而阴性样品在相应的位置上无斑点。结果见图2。
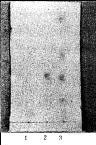
图-2 黄芩薄层色谱图
1. 阴性样品 2. 黄芩苷对照品 3. 样品
-
+关注
私聊
-
yhl-87_
第15楼2009/11/06
薄层色谱法应用系列讲座(14) (下)
4 小儿清热速崩片中麻黄碱含量的HPLC法测定
4.1 色谱条件
色谱柱:Agilent - C18柱(4.6 mm×150 mm×5 μm);流动相:O.1%磷酸溶液(100 mL中加三乙胺0.1 mL)一乙腈(99∶3);检测波长:205nm;流速:0.8 mL·min-1;进样量:l0μL。
4.2 线性范围的考察
精密称取盐酸麻黄碱对照品28 mg,置100 mL量瓶中,加水使溶解并稀释至刻度,摇匀,得含盐酸麻黄碱280 μg·ml-l的溶液。分别精密吸取1 mL以水稀释至2,5,10,25,50 mL摇匀,得盐酸麻黄碱浓度分别为280, 140,56,28,11.2,5.6 μg·mL-l的对照品溶液。分别吸取不同浓度的对照品溶液10μL,注入高效液相色谱仪,测得峰面积,以盐酸麻黄碱量(μg)为横坐标,峰面积为纵坐标绘制标准曲线,得回归方程为:
Y=2.99×103X-15.9 r=0.9999线性范围为0.06~2.8 μg。
4.3 供试品溶液的制备及测定
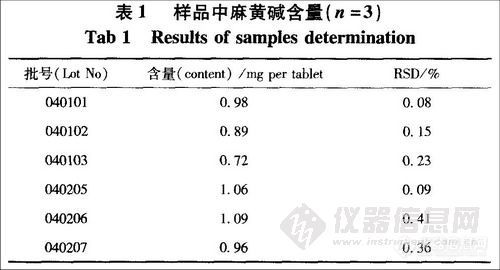
取本品20片称定,研细,取1.5g,精密称定,置半微量定氮瓶中,加水与40%氢氧化钠溶液各10 mL,以盐酸溶液(1→20)5 mL作为吸收液,加热,蒸馏,收集馏出液至75mL时停止,定容至100 mL,摇匀,滤过,即得供试品溶液。精密吸取供试品溶液及28μg·ml-l对照品溶液各10μL进样测定,以外标法计算含量,1 mg盐酸麻黄碱相当于0.819 mg麻黄碱,测定结果见表1。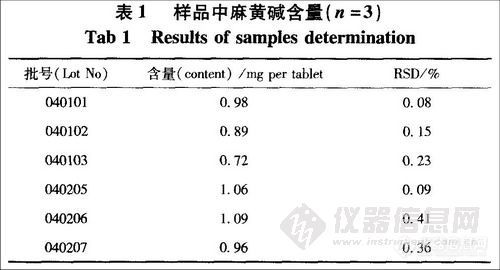
4.4 阴性对照试验
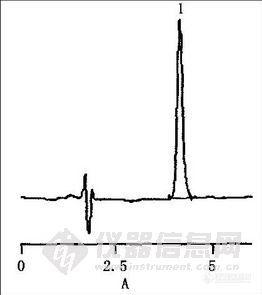
照小儿清热速崩片处方制得缺麻黄药材的阴性样品,按“4.3”项制各阴性供试品溶液,取10μL进样。阴性样品图谱中未见盐酸麻黄碱色谱峰处有假阳性峰,可见样品中各组分对盐酸麻黄碱含量测定无干扰(见图3)。
A. 为对照品 B. 为样品 C. 为阴性样品
4,5 精密度试验
精密吸取同一供试品溶液,重复进样6次,测定峰面积,RSD=0.67%。
4.6 重复性试验
取同一批号(040205)样品6份,分别按“4,3”项下操作,计算得平均每片麻黄碱含量为0.89 mg·片l, RSD=0.58%。
4.7 稳定性试验
同一供试品溶液放置不同时间(0,2,4,8,24 h)后进样测定峰面积,其RSD=0.49%,表明本法处理的供试品溶液在24 h内稳定性良好。
4.8 加样回收试验
取已知含量的样品(批号040205)约0.75 g(平行6份),分别置半微量定氮装置中,精确加人浓度为0.668 mg·ml-1的对照品溶液1 mL,按“4.3”项下制备所需溶液并测定,计算平均回收率为99.8%,RSD=0.56%。
5 讨论
5.1 黄芩鉴别方法选择
中国药典法[l]报道小儿清热止咳口服液黄芩鉴别方法为化学比色法,由于小儿清热速崩片辅料多,颜色变化不明显,因此我们选择了薄层色谱法,此法斑点清晰。
5.2 含量测定中波长的选择
文献[2,3]报道盐酸麻黄碱含量测定的检测波长有210 nm、245 nm,本实验测定了盐酸麻黄碱对照品溶液的紫外吸收图谱,最大吸收波长为205.3 nm,故选择205 nm作为检测波长。
5.3 含量测定中样品处理方法的选择
中国药典法[1]报道小儿清热止咳口服液麻黄碱鉴别方法中样品前处理选用萃取法,此操作过程中易乳化,选用此方法做前处理测含量,回收率仅达到85%,且杂质峰很多。麻黄碱为小分子生物碱,具有挥发性,可用蒸馏法提取。故选用蒸馏法提取小儿清热速崩片中麻黄碱,作为样品前处理方法,杂质峰少,回收率好。
5.4 含量测定中流动相的选择
曾试用了3种流动相,并调整了流动相中各组分的比例:
(1)0.08mol·L-1磷酸二氢钾一乙腈(90:l0),用三乙胺调pH=6.8);
(2)0.l%磷酸一甲醇(97:3含三乙胺0.1 mL)作为流动相;
(3)0.1%磷酸(100 ml中加三乙胺0.1 mL)一乙腈(97:3)作为流动相。结果发现0,1%磷酸溶液(100 mL中加三乙胺0.l mL)一乙腈(97:3)作流动相,样品中盐酸麻黄碱色谱峰与其他峰无干扰,可获得较好分离效果其峰形也最好。
参考文献
1 ChP(中国药典)2000 .Vol I(一部):373
2 YANG Lei(杨蕾),HE Jian-hua(贺建华),HE Xia-qiu(何夏秋), et al .Determination of ephedrine hydrochoride in Xuanfei Qutan oral liquor by HPLC assay(HPLC测定宣肺祛痰口服液中盐酸麻黄碱)Chin Pharm J(中国药学杂志),2000,35(8):548
3 LIU Gui - zhong(刘贵中)Determination of ephedrine In Biyan Diji by HPLC assay(高效液相色谱法测定鼻炎滴剂中麻黄碱的含量)Chin J Pharm Anal(药物分析杂志),1998,18(5):341
-
+关注
私聊
-
yhl-87_
第16楼2009/11/06
薄层色谱法应用系列讲座(15)-氨基酸结构与保留关系的研究
反向传播人工神经网络用于薄层色谱中
氨基酸的结构与保留关系的研究
摘要 在测定、收集和计算出一组氨基酸的拓扑指数和各种理化参数之后,再通过相关分析选择其中最有代表性的几个参数作为反向传播人工神经网络的输入参数,用于正相薄层色谱中氨基酸保留规律的研究。结果表明·氨基酸的色谱保留值与其结构之间呈现较强的非线性关系,采用人工神经网络方法比用多元线性回归方法能够更精确地描述这种关系。
1、 前言
人工神经网络方法中,用得较多的是反向传播神经网络方法⑴。由于这一方法能较好地处理非线性体系,因而已逐步得到广泛使用。而对物质的结构与色谱保留值之间关系的研究也一直是色谱工作者感兴趣的课题。氨基酸是人们研究得比较多的一类重要的化合物。薄层色谱法很早就被用于氨基酸的研究。但由于氨基酸虽然在命名上是一类,而在结构上差别却较大,加上薄层色谱中影响因素较多,氨基酸的薄层色谱保留与它们的结构之间的关系往往呈现较强的非线性,因此,采用传统的线性回归方法描述二者的关系一直都不太理想。
本文由中国科学院大连化学物理研究所的王岳松、张军和林乐明研究员们共同完成,他们不仅测定、收集和计算了氨基酸大量的拓扑指数和理化参数,并对这些参数进行相关分析,利用相关分析从中优选出几个代表性的参数,再将这几个参数作为人工神经网络的输入参数。这样,既避免了人工神经网络对输入参数选择的盲目性,也使人工神经网络参数具有较明确的物理意义。同时还采用了多元线性回归方法和人工神经网络方法,研究了正相薄层色谱中氨基酸保留值与其结构之间的关系,得到了单用回归分析所不可能达到的、比较精确的结果。下面具体介绍他们的论文。
2、 实验部分
2-1仪器与试剂
CAMAGⅡ型薄层色谱扫描仪/CATs3.17工作站(瑞士CAMAG公司),PBQ型自动薄层铺板器(重庆新力试验电器厂),超声波发生器(CSF一1A型,上海超声波仪器厂),xY双底展开缸(上海信谊仪器厂),Linomat.Ⅳ喷样仪(瑞士CAMAG公司)。硅胶H(青岛海洋化工厂)。除了酪氨酸(Tyr)用体积分数为o.1%的HC1溶液配制外,其它氨基酸均用体积分数为1o%的丙醇配制,质量浓度约为1g/L。所有试剂均为分析纯。
2-2色谱条件
实验室自涂板:m(硅胶H):y(10g/L的CMC)一1g:2.5mI,,玻璃载板,层厚度o.25mm,110℃下活化0.5h,取出,放入干燥器中备用。在双底展开缸中上行展开,展距8.5cm,待板干后,喷洒茚三酮一硝酸铜溶液,并在105℃下加热1.5~2min。在460nm(钨灯,反射吸收模式)下扫描。
3、 结果与讨论
3-1 15种氨基酸的有关数据
氨基酸可分为脂肪族、芳族和杂环族,脂肪族氨基酸又分为中性、酸性、碱性、含羟基和含硫等5类。在常见的23种氨基酸中,我们选择了除含硫氨基酸外的15种更常见的氨基酸(其中,中性4种、酸性3种、碱性2种、含羟基2种、芳族2种、杂环2种)作为研究对象,以便使之具有足够的代表性。15种氨基酸的品(比移值)实验值列于表1中,Randic:分子连接性指数(Xoυ,X1υ)⑵⑶⑷、Pyka提出的两个指数(A,0B)⑸、Gutman指数(M)⑹、Wiener指数(W)⑺和Balaban指数(D J)⑻列于表2,物理化学参数则列于表3。表3中Mw表示相对分子质量,Vw,Aw ,MR分别代表范德华体积、表面积和摩尔折光率,N (%)是分子中氮的质量分数,d.P.和PI分别是分解温度和等当点。
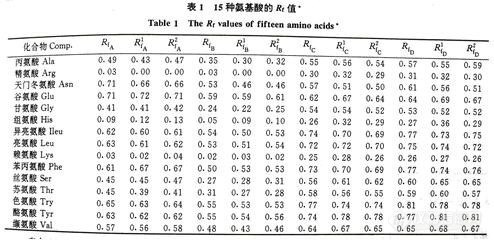
表中,展开剂:RfA ,V(乙醇):V(水) = 4:1;RfB,V(乙醇):V(水) = 8:1,Rfc,V(丙醇):V(醋酸):V(水) = 3:1:1; RfD,V(丙醇):V(醋酸):V(水) = 6:2:1。
上标 1:RfA和RfB是以Asn,GIu,Gly,His,I1eu,Lys,Phe,Ser,Try’Tyr等10种氨基酸作为训练集,所有15种氨基酸作为预测集;Rfc和RfD是以Arg,Asn,GIu,Gly,Ileu,Lys,Phe,Ser,Try,Tyr等10种氨基酸作为训练集,所有15种氨基酸作为预测集。
上标 2:以所有15种氨基酸为训练集。
表-2 15种氨基酸的拓扑指数 7425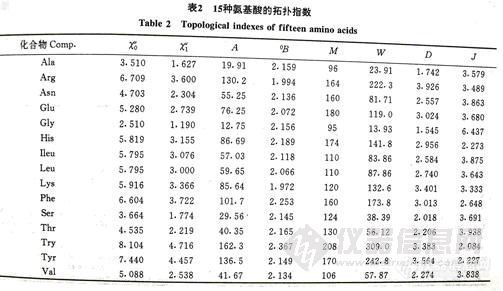
表-3 15种氨基酸的理化参数 7426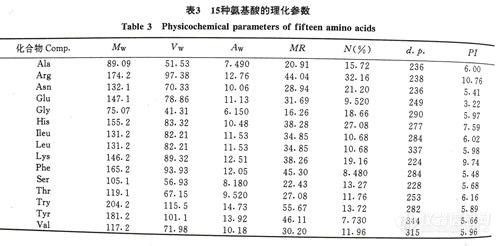
3-2 各种参数的相关分析和人工神经网络处理
从统计分析的角度来说,样本数一般应该是变量的5倍以上。同时,为了用最少的变量代表所有参数中尽可能多的信息,我们首先对所有的参数通过相关分析进行筛选。相关系数矩阵,列于表4。
显然,当两个参数的相关系数的绝对值接近1时,用它们来表达分子结构与色谱保留值之间关系的效果应该是基本相同的。当多个参数与同一个参数的相关系数的绝对值都为1时,它们的表达效果将是叠加的。因此,我们可以将与同一参数的相关系数接近1的这些参数划为一类。根据这一原则,参照表4所列出的相关系数,基本上可以将所有参数分为3类:① Mw ,X oυ, X1υ, A,W,D,J,Vw,Aw,MR;② B,M;③N (%),d.p.,PI。
从每一类中各选出1个参数,再经对不同的溶剂系统进行优化后,选出不同的3个参数参与下一步的神经网络处理。例如,对乙醇-水系统,选用PI,M,Aw,参数;对于正丙醇-醋酸系统,选用PI,M,W参数。这里,PI反映着分子的酸碱性,M及W代表着分子的拓扑结构和分子的大小,Aw也反映着分子的大小。
但是,当将参数输入神经网络时,由于作为传递函数的Sigmoid函数对于大于1的数反应不灵敏,因而输入时取PI,Aw的百分之一和M,W的千分之一。我们采用的是3-5-1型网络,即输入层设3个输入节点、隐含层设5个节点和输出层设1个节点。神经网络运行中,学习次数为80 000次左右,未发现超拟合现象。学习速率和动量因子经优化取为0.6。通过人工神经网络计算得到的Rf值列在表-1中。
由于所选取的10个作为训练集的氨基酸代表着要预测的所有的氨基酸类型,因此表-1中的预测结果反映着神经网络系统的预测能力。对预测Rf值与实验得到的Rf值进行相关分析,所得统计分析结果列于表-5。
表-4 15种参数的相关系数矩阵 7427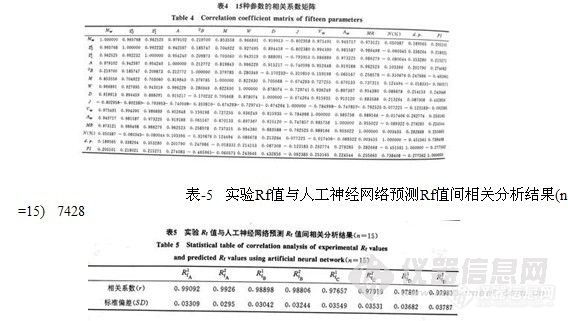
从表-5可以看出,用人工神经网络方法对氨基酸的Rf值的预测结果令人满意,与实验值之间的相关系数均在0.98左右。同时也可看出,用10个氨基酸作为训练集与用全体作为训练集的预测结果极为接近。这说明,用人工神经网络预测氨基酸的薄层色谱保留值具有更高的精确度和可靠性。
参考文献
1 Rumelhart D E,Hinton G E,Willams R J.Learninginternal representations by error propagation.In:Rumelhart D E,McClelland J L eds.Microstructures of Cognition.
Vo1.1.Cambridge:NIT Press,1986.318
2 Kier L B,Hall L H,Murray W J et a1.J Pharm Sci,1975,64:1971—1974
3 Randic M.J Am Chem Soc,1975,97:6609—6615
4 Kier L B。Hall L H.Molecular connectivity in strut ture-activity analysis.New York:RSP,1986.25
5 Pyka A.J Planar Chromatogr,1991,4:316-318
6 Gutman I,Ruseic B,Trinajstic N.J Chem Phys,1975,62:3399-3405
7 Wiener H.J Am Chem Soc,1947,69:17—20
8 Balaban A T.Pure Appl Chem,1983,55:199—202
-
+关注
私聊
-
yhl-87_
第17楼2009/11/06
薄层色谱法应用系列讲座(16)-庆大霉素及其相关物质
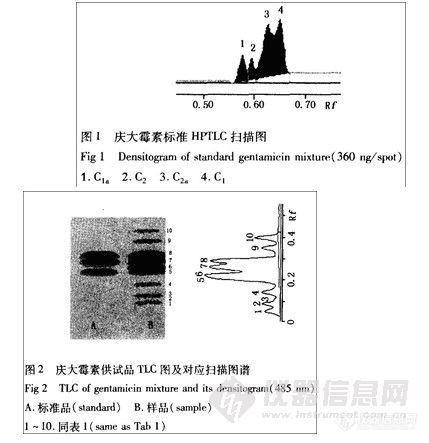
HPTLC法测定庆大霉素组分及其相关物质
摘要 目的:建立测定庆大霉素组分及其相关物质的HPTLC法。
方法:用高效薄层色谱法测定庆大霉素组分及其相关物质。CAMAG全自动薄层色谱仪;薄层板采用 MERCK高效硅胶G板,展开剂为氯仿一甲醇-25%氨水(5:7:6),测定波长为485nm。
结果:庆大霉素在3.98~39.8μg·mL-1范围内各组分呈良好的线性关系(r >0.99);检测限为 ng级。庆大霉素注射液中组分及其相关小组分之间分离度良好。
结论:该方法简单、快速,灵敏度高,适用于庆大霉素组分中相关物质的分离测定。
前言
庆大霉素(gentamtcm,GM)是由小单孢菌属(Micrornonospora purpurea)发酵产生的一组结构相近的多组分氨基糖苷类抗生素[1]。根据其分子结构的差异可分为庆大霉素C与庆大霉素A两组:C组包括C1a,C2,C2a,C1等;A组包括A,A1~A4,B,B1和X2等[2]。C组庆大霉素为杀菌力较强的广谱抗生素,目前临床中广泛使用,其中C1a,C2,C2a,C1之和通常占90%以上;A组庆大霉素抗菌活性弱但对原虫、蠕虫有很好的效果[3]。在生产过程中,因发酵菌种的变异及纯化工艺的变化,庆大霉素的组成及比例常发生变化,因此对庆大霉素组分的控制成为其质控最重要的环节之一。
目前,各国药典均采用HPLC法对庆大霉素中的C组分进行控制,, USP 28版和BP 2004采用柱前衍生化方法检测[4,5];中国药典2005年版则采用ELSD法检测[6]。虽然2种测定方法对庆大霉素的4个C组分均能达到较好的控制,但对其中小组分的测定却存在明显的不足。在HPLC衍生化法中,很难判断检测到的杂质峰究竟是样品本身含有的小组分还是衍生化反应的副产物;而HPLC - ELSD方法的检测灵敏度较低。此外,文献报道的庆大霉素C组分的测定方法还有核磁共振法(NMR)[7]和薄层色谱(TLC)法[8,9]。由于NMR仪的操作及维护需要较高的专业背景,大大限制了NMR的普及和应用。TLC法虽然是最早被开发用于分析庆大霉素C组分的方法,但长期以来传统TLC法在药物分析领域只是作为一种半定量的分析手段,其测定结果易受多种因素的影响。由于庆大霉素组分间结构相近,组成复杂,使得TLC测定难以达到较好的分离度和重复性。随着现代薄层色谱技术的发展和TLC法自动化程度的提高,高效薄层色谱(HPTLC)已经越来越多地运用于药品质量分析和控制领域[10,11],并表现出独特的优点。
本文由杨利红①②③、胡昌勤②、刘文英①(①中国药科大学;②中国药品生物制品检定所;③黑龙江省药品检验所)尝试建立了高效薄层色谱法(HPTLC)对庆大霉素组分进行分析测定,利用自动化控制程序对色谱条件进行优化,并与HPLC-ELSD方法进行对比,以达到准确分析庆大霉素组分及相关物质的目的。现全文介绍如下:
实验
1 仪器与试药
CAMAG全自动薄层色谱仪(包括全自动点样装置ATS4;全自动薄层展开系统AMD2;自动薄层扫描仪SCANNER3;全自动数据成像系统;平面色谱工作站winCATS(VI.2.1,CAMAG),Switzerland;高效硅胶G、GF254薄层板20 cm×l0 cm(Merck公司);国产高效硅胶G、GF254玻璃板(青岛海洋化工厂);普通硅胶G薄层板(Merck公司)。美国Alltech公司HPLC - ELSD仪(ELSD 2000)。
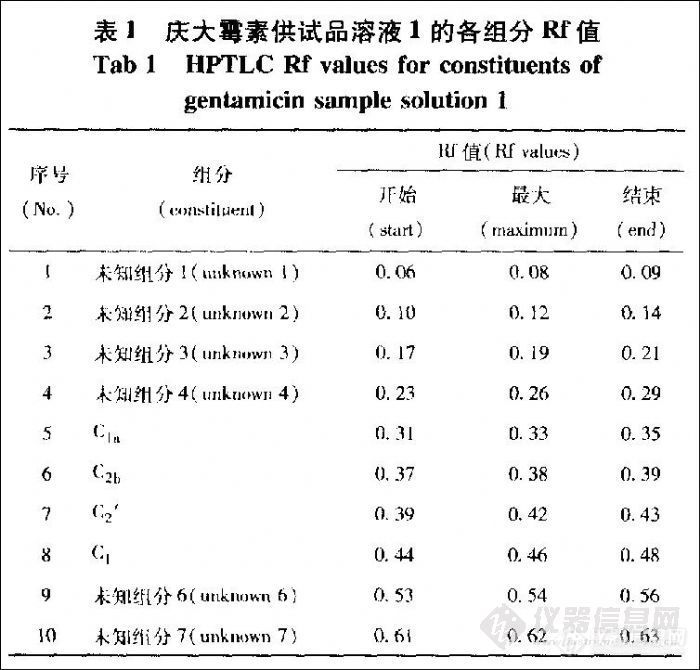
庆大霉素C组分标准品(批号:130326 -200314,每1 mg 638单位;其中C1a 32.9%,C2 28.8%,C2a 19.5%,C1 19.7%),庆大霉素C1a,C2,C2a,C1工作对照品;小诺霉素标准品(批号3429402,每l mg 577单位);庆大霉素原料(编号为HHAC200500166);硫酸庆大霉素注射液(2 mL:8万单位)2批(编号分别为HCAC200502140和HCAC200502141);均由中国药品生物制品检定所提供。所用化学试剂均为分析纯(美国Dima公司)。
2 方法与结果
2-1 HPTLC方法的建立
2-1-1 标准品溶液的配制
精密称取庆大霉素标准品适量,用水制成0.2 mg·mL-1(标1)和2 mg·mL-1(标2)2种浓度的溶液;另精密称取庆大霉素C1a 对照品适量,用水制成0.4 mg·mL-1(标3)浓度的溶液;另分别取庆大霉素各工作对照品配制成一定浓度的定位对照溶液。
2-1-2 供试品溶液的配制
精密称取硫酸庆大霉素原料粉末适量,用水溶解并制成1 mg·mL-1的供试品溶液1;另精密量取硫酸庆大霉素注射液按标示量加水制成1 mg·mL-1的供试品溶液2。
2-1-3 标准曲线的绘制
照薄层色谱法[6]试验,分别吸取标1溶液1,3,6,8μL和标2溶液1,3,6,8,10μL,点样于同一高效硅胶G薄层板上;每一体积平行点样4次;斑点设定为2 μm的条带状。以各体积斑点的平均峰面积对应各斑点浓度作标准曲线。
2-1-4 层析方法
采用自动展开装置,以氯仿-甲醇-25%氨水(5:7:6)混合I h后取下层液为展开剂;展开75 mm;干燥时间l0 min;取出后,挥散氨气味,放人碘熏缸中8~10 min,取出。
2-1-5 扫描条件的确定
标准扫描:光狭缝6.00mm×0.40 mm,扫描分辨率50 μm/step,灯D2&W,波长485 nm;光谱扫描:对标准扫描图谱进行处理后进行各指定峰的光谱扫描;灯D2&W;扫描波长200~700 nm;测定模式Absorption;光谱扫描速度l0 nm·s-1。
2-2 HPLC - ELSD法 [6] 参照中国药典2005年版硫酸庆大霉素品种检查项下进行操作。

-
+关注
私聊
-
yhl-87_
第18楼2009/11/06
薄层色谱法应用系列讲座(16)(下)-庆大霉素及其相关物质
3 HPTLC方法的评价
3-1 专属性试验
分别取庆大霉素标2溶液及供试品溶液1各2 μL,照“2-l”项下方法操作。用“2-I”项下庆大霉素各主组分定位对照溶液进行TLC定位,并经HPLC - ELSD法验证。较低浓度时庆大霉素标准品溶液HPTLC扫描图谱中4个组分能较好分离,见图1;但其斑点浓度超过726 ng时,C2和C2a的TLC色谱峰(C2+C2a即C2′)重合。庆大霉素供试品TLC斑点及其对应扫描图谱见图2,各组分的Rf值见表1(C2b由小诺霉素标准品定位)。图谱显示庆大霉素各组分及其相关物质之间能够较好地分离。

3-2 线性关系考察
照“2-1-3”项下方法操作,庆大霉素标准溶液在3.98~39.8μg·mL-1范围内,各纽分的浓度与其斑点的面积积分值均呈良好线性关系。组分C1a,C2’,C1的线性方程分别(n=6)为:
y=593.39X+3608.5 r=0.9982
y=593.99X+209 1.5 r=0.9971
y=563.93X+3555.2 r=0.9931
其中C2’和C1标准曲线的响应因子差别较大。采用t检验法考察2个方程问的差异显著性,结果表明两者问无显著性差异。
3-3 重复性试验
选取线性考察中庆大霉素标准品溶液测定的数据,以各组分的3个不同浓度的4次测量峰面积计算,C1a,C2’,C1的RSD分别为3.2%,4.2%,2.6%。
3-4 溶液稳定性考察
取庆大霉素标2溶液6μL,在同一薄层板上连续3 d分别点样6次,以各斑点的峰面积计算C1a,C2’,C1的RSD分别为5.3%,2.6%,4.4%。
3-5 检测限和定量限
信噪比为3:1时C1a,C2,C2a,C1的最低检测浓度分别为80,95,95,70 ng;信噪比为l0∶1时C1a,C2’,C1的最低定量限分别为382,390,279 ng;另取标2和标3溶液,按“3-8”项下方法操作,以标2溶液为对照计算标3的量,准确度为I06.l%;6次点样的精密度为5.6%。
3-6 加样回收试验
准确量取已知含量的庆大霉素注射液I.25 mL(相当于50 mg)于100 mL量瓶中,再加人精密称量的庆大霉素标准品约50 mg,用水溶解并稀释至刻度,按“3-8”项下方法操作,计算,庆大霉素C1a,C2’,C1的回收率(n=6)分别为104.3%,98.2%,96.8%;RSD 分别为 4.1%,3.2%,4.2%。
3-7 粗放耐用性试验
3-7-1 TLC板的适用性考察
考察Merck公司和国内生产企业的不同种类的TLC板对试验的适用性,发现用Merck生产的硅胶G60或GF254 HPTLC板对庆大霉素各组分具有较好的分离;庆大霉素注射液中分离出13个组分;Merck普通TLC玻璃板或铝板及国内生产的HPTLC板只能分离出l0个组分;其中国产板的组分斑点还易出现拖尾现象。
3-7-2 温湿度对实验的影响
考察结果表明温度在18~30℃、相对湿度在27%~70%范围内实验结果基本一致。
3-7-3 展开距离及装置的影响 采用高效板展开50 mm以上时即可对庆大霉素各组分进行分离;手动与自动展开结果没有明显改变。
3-7-4 碘蒸气显色时间及放置时间的影响
一般以显色8~15 min薄层板面呈现清晰样品斑点后,取出,立即测定为宜。
3-8 样品测定
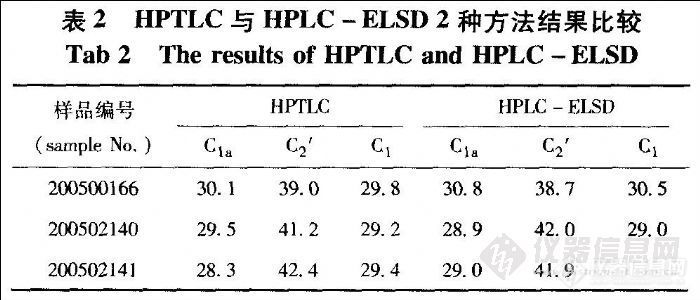
取2 mg·mL-1的庆大霉素标准品溶液,分别点样1,2,3,4,5 μL,每个体积重复点样4次;另取供试品溶液1和2各2 μL点样于同一TLC板上,各平行点样6次。做标准曲线并将每份供试品6个斑点的平均峰面积代入曲线方程,计算得供试品中庆大霉素各组分的百分含量。各批中C1a.组分的RSD最大,分别为3.5%,2.6%,3.8%,n=6。同时按HPLC - ELSD法测定各组分含量。结果见表2。
4 讨论
4-1 展开系统的确定
传统TLC测定庆大霉素采用氯仿一甲醇-氨水(1:1:1)展开系统,因庆大霉素各组分结构相近,其比移值(Rf)相差较小,且其具有碱性结构使得斑点拖尾严重,各组分的分离度均很难达到要求。考虑增加其展开剂的极性有助于增加Rf值,但水相比例太大其组分斑点又易扩散,最终也影响分离。因此对不同比例的展开系统进行比较,最终确定氯仿一甲醇-25%氨水(5:7:6)作为展开剂。
4-2 扫描波长的确定
薄层板展开显色后在200~700 nm范围内扫描,可以在线提取各组分斑点的光谱,确定其最大吸收波长;各组分均在300 nm附近有最大吸收。但因TLC板经碘熏显色后背景颜色较深,在300 nm有较高的基线背景,影响了小组分的检出和主组分的定量。经试验后确定测量波长为485 nm。
4-3 响应因子一致性考察
对“3-2”中三组分的回归方程进行比较,对其中差别较大的方程其回归系数和截距作t检验。结果其主组分在HPTLC—光密度扫描法中的响应值基本一致。这与其响应因子在ELSD中的一致性相似。
4-4 显色方法及时间的影响
因碘具有升华性,因此碘熏显色后应立即测定。为能使所显的颜色在一段时间内稳定,可用一块大小相同的干净玻璃板,将显色后的薄层盖住,周围用透明胶带密封,使其与外界隔绝。另外,如果显色时间过长,背景颜色较深,就会造成较大的测定误差。我们可以利用碘的升华性,将薄层板暴露在空气中放置一会儿再测定或者等碘升华后,颜色逐渐褪去,再次用碘或其他方法显色。轨道扫描从薄层板一侧向另一侧逐一进行,需一定时间,为消除扫描过程中碘蒸气挥发造成的误差,同一块薄层板上点样数目不宜过多。另外,也可将对照溶液分别点样于薄层板两侧。
4-5 HPTLC法和HPLC-ELSD法比较
将HPTLC和HPLC-ELSD 2种方法测定结果进行比较(表2),并选择差距较大的C1a组分的计算结果进行双因素方差分析,表明HPTLC与HPLC-ELSD 2种方法定量测定庆大霉素主组分其结果在统计学95%(P =0.05)置信区间没有显著性差异。
薄层色谱法应用系列讲座(16)-庆大霉素及其相关物质
5 结论
比较实验结果可以看出,定量测定庆大霉素各组分时HPTLC中的C2和C2a,斑点重合,只能对其合并计算;另外由于庆大霉素结构的特点,HPTLC法采用碘熏显色光密度扫描对其组分进行定量,斑点之间易产生干扰,造成测量误差增加。但同时也不难看出HPTLC法具有更高的灵敏度(其定量限与ELSD法相比低近7倍);在检测浓度低10倍的情况下HPTLC可检测出庆大霉素供试品中至少6种相关小组分,而ELSD只检到5种。另外HPTLC还具有多通道测定的特点,对于多个未知样品的分离测定更方便、快捷和直观,并可以在线监测任一组分的UV、可见或荧光光谱。因此在定性分析庆大霉素相关物质方面HPTLC具有更明显的优势。HPTLC的正相系统也是对HPLC反相色谱系统很好的补充。其微小组分在HPTLC中的定位和定性将在日后工作中进一步研究。
HPTLC法作为一种日益完善的分析手段,已具有了较高的精密度和准确性。在进一步优化色谱条件改善分离的前提下,HPTLC完全可以成为测定庆大霉素主组分的一种定量方法;并在分析测定庆大霉素相关物质方面具有较明显的优势。
-
+关注
私聊
-
yhl-87_
第19楼2009/11/06
薄层色谱法应用系列讲座(17)食品袋中酞酸酯含量
薄层色谱扫描法测定塑料食品袋中酞酸酯的含量
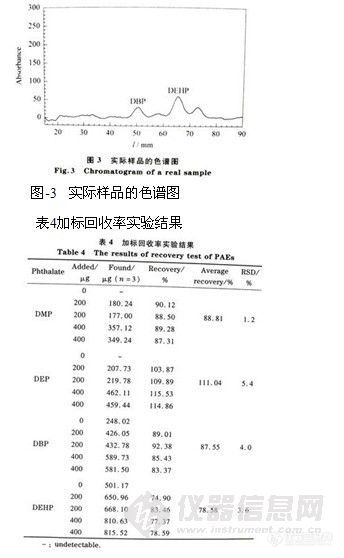
摘要 建立了采用薄层色谱扫描测定塑料食品袋中4种酞酸酯、酞酸二甲酯(DMP)、酞酸二乙酯(DEP)、酞酸二丁酯(DBP)和酞酸二-2-乙基己基酯(DEHP)的方法。经粉碎的样品先用乙醇浸泡24 h,然后超声提取,经0.45μm滤膜过滤。点样在以丙酮处理过的硅胶G板上,以乙酸乙酯-无水乙醚-异辛烷 (体积比为1:4:15)为展开剂,以双波长反射吸收飞点扫描测定(λs=275nm,λR=340nm),外标法定量。该法的线性关系较好,DMP、DEP、DBP和DEHP的检出限分别为2.1、2.4、3.4和4.0ng,混合标准品在同一薄层板上的峰面积的相对标准偏差(RSD)为2.8%~3-5%,4种酞酸酯的样品加标回收率为78.58%~111.04%。该方法样品用量少,前处理简单,分离效果好,可用于塑料袋中4种酞酸酯的同时测定。经与气相色谱法的分析结果比较,两种方法对实际样品的分析结果接近。
本文由首都师范大学化学系的陈惠、汪瑗和朱若华老师们采用薄层色谱方法来测定DMP、DEP、DBP和DEHP,在20min内能够很好地分离4种化合物,可用于塑料食品袋中PAEs的测定,下面介绍他们的实验和结果。
1 前言
酞酸酯(phthalic acid esters,PAEs)是邻苯二甲酸酯类物质,含有较弱的雌激素成分,可影响生物体的内分泌、致突变、致畸和癌细胞增殖,是一类环境激素。可通过呼吸、饮食和皮肤接触进人人体内,对人体健康造成危害⑴。
PAEs主要用作塑料增塑剂,广泛应用于建筑材料如塑料地板、壁纸、涂料和汽车座椅、椅套以及衣服、玩具、化妆品、香料、医疗设备等的生产过程中。由于PAEs与塑料基质之间没有形成化学键,而是以氢键和范德华力连接,故接触到合适的有机溶剂便会溶解出来。常用的商品化酞酸酯约14种,其中6种被美国环保局(EPA)列入129种重点控制的污染物名单,分别是酞酸二甲酯(DMP)、酞酸二乙酯(DEP)、酞酸二丁酯(DBP)、酞酸二-2-乙基己基酯(DEHP)、酞酸二辛酯(DOP)和酞酸丁基苄基酯(BBP);8种被列入世界野生动物基金会 (wwF)的环境激素名单。我国也将PAEs中的DMP、DBP和DOP列入优先控制的污染物黑名单。随着塑料工业的发展,塑料制品的大量使用已使酞酸酯成为环境中无所不在的污染物。
PAEs的测定方法有分光光度法、气相色谱法(GC)⑵⑶和高效液相色谱法(HPLC)⑷。分光光度法只限于酞酸酯总量的测定;色谱法选择性强,可单独测定各种酞酸酯的含量⑸。EPA的标准方法是气相色谱-电子捕获检测-质谱法(GC-ECD-MS)⑹。,我国拟发布的标准方法是正相HPLC,主要是分析环境样品如土壤⑺、水样⑻⑼、大气⑽中的PAEs。目前还没有标准及成熟的技术检测塑料产品中的PAEs。薄层色谱是应用较为广泛的简便分析方法,但用薄层色谱来进行PAEs的定量检测尚未见报道。
2 实验部分
1.1 仪器与试剂
CX-250型超声波清洗机、Densitometer CD 60高效薄层色谱扫描仪 (德国) 、微量进样器 (上海医用激光仪器厂)、硅胶G板 (青岛海洋化工厂)。
DMP、DEP、DBP、DEHP由北京市理化分析测试中心提供。异辛烷、乙酸乙酯、无水乙醚、甲醇、无水乙醇均为分析纯。
1.2 标准溶液的配制
准确称取DMP、DEP、DBP、DEHP标准品各0.1000g,分别用甲醇稀释定容至l0ml,配制成l0.000g/L的单标储备液。分别移取上述4种单标储备液各l mL,用甲醇定容至10mL,配制成1000mg/L的混合标准储备液。再用甲醇将上述溶液逐级稀释为500mg/L和50mg/L备用。
1.3 样品处理
本文所用样品为本校食堂常用就餐塑料食品袋。精确称取约0.5g的塑料食品袋,剪成碎片放入具塞三角瓶中,用20mL无水乙醇浸泡24 h,超声提取3次,每次15min。然后过0.45μm滤膜,水浴蒸发浓缩,用无水乙醇定容至15mL备用。
1.4 分离测定
定量吸取标准品混合液和样品提取液,点样于同一块硅胶G薄层板上,以乙酸乙酯-无水乙醚-异辛烷(体积比为1:4:15)为展开剂,饱和30min,直立上行展开70mm,挥干溶剂,将薄层板置于薄层扫描仪的工作台上,在测定波长275nm、参比波长340nm的条件下进行双波长反射吸收飞点扫描测定。根据DMP、DEP、DBP和DEHP标准品斑点的Rf值定性,外标法定量。
1.5 气相色谱分析
Agilent 6890N气相色谱仪;载气为N2 ;流量为0.8mL/min ;色谱柱为HP-5MS毛细管柱 (30.0m×0.32mm×0.25μm);进样方式为手动,不分流;进样量为1~5μL;进样口温度为250℃;检测器为火焰电离检测器(FID),检测器温为300℃;柱压为41.86kPa.(恒压);升温程序:150℃(3min)以15℃/min升至300℃(4min)。以保留时间定性,峰面积外标法定量。
3 结果与讨论
2.1 薄层板的优化
经高效薄层扫描仪扫描发现,空白薄层板的背景值较高,且不均匀,因此薄层板需先在110℃烘箱中活化1 h,再用有机溶剂进行展开处理,将薄层板上可能的杂质带到板的前沿,以降低板本身的背景和杂质的干扰。分别考察了三氯甲烷、丙酮和所选用的展开剂进行处理,结果表明经丙酮处理过的薄层板效果最好,因此本实验中均使用经丙酮处理过的薄层板,处理后的薄层板置于烘箱中75℃烘干,再置于干燥器中备用。
2.2 扫描测定波长的确定
在经处理的薄层板上用1μL微量注射器按间隔15mm的距离,均匀点上DMP、DEP、DBP和DEHP标准储备液各0.5μL。晾干后,用薄层扫描仪进行反射吸收光谱扫描。DMP、DEP、DBP和DEHP的反射吸收光谱见图-1。由图-l可见,PAEs在230nm和275nm、处有紫外吸收,但是在230nm处的背景值较高,所以采用275nm为测定波长,340nm为参比波长。
2.3 展开剂的选择
以乙酸乙酯为基体,加入其他试剂,适当调节展开剂的极性。对酞酸酯标准液展开分别采用了3种展开剂:① 乙酸乙酯-异辛烷 (1:9,体积比,下同),② 乙酸乙酯-无水乙醚-异辛烷(1:4:15),③ 乙酸乙酯-正已烷(15:85),
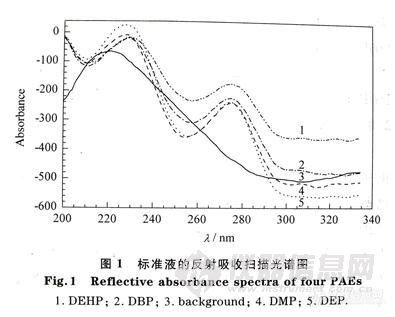
图-1标准液的反射吸收扫描光谱图
1. DEHP;2. DBP;3.background:4.DMP:5.DEP.
结果见表-l。展开剂①可分离4种PAEs,展开需40min以上;展开剂②采用对酞酸酯有更好亲和力的乙醚偶极试剂,溶剂极性适中,20min内可展开,且可较好地分离4种PAEs (见图2),干扰少,峰形好,各物质的Rf值均落在0.30至0.65之间,在定量分析范围内。展开剂③可分离4种PAEs,但Rf值偏大,峰拖尾。因此最佳展开剂为②,即乙酸乙酯-无水乙醚-异辛烷(1:4:15)。
表-1不同展开剂条件下酞酸酯的比移值(n=3)
-
+关注
私聊
-
yhl-87_
第20楼2009/11/06
薄层色谱法应用系列讲座(17)(下)食品袋中酞酸酯含量
2.4 线性范围和检测限
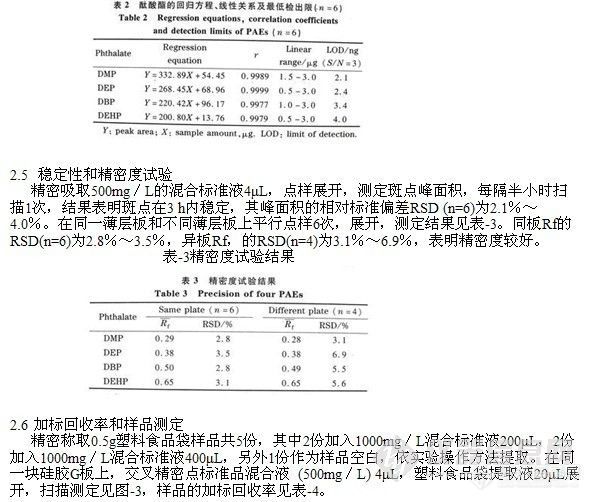
精密吸取酞酸酯混合标准品溶液 (500mg/L) 1.0,2.0,3.0,4.0,5.0,6.0μL,分别点样于同一硅胶G薄层板上,按“1.4”节方法进行展开测定,以峰面积的积分值为纵坐标,标准品的点样量 (μg)为横坐标,绘制标准曲线,其回归方程、相关系数、线性范围和最低检出限见表-2。
表-2酞酸酯的回归方程、线性关系及最低检出限(n=6)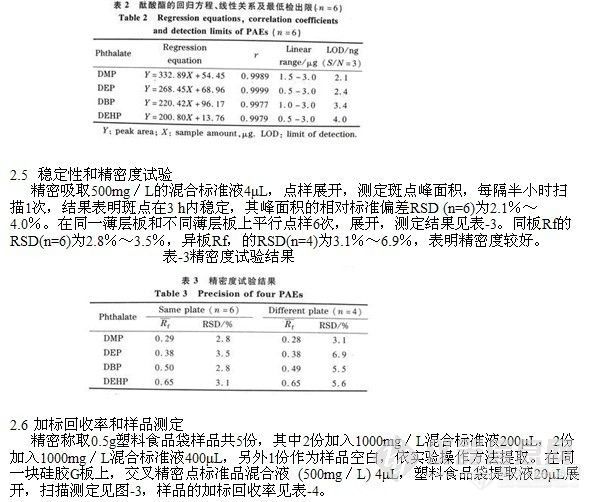

由空白样品的实验结果可计算出该塑料食品袋中DBP和DEHP的含量分别为0.05%和0.10% (质量分数),这与我国酞酸酯生产应用中以DBP和DEHP为主 (>90%)有关。
2.7 GC分析结果
在“1.5”节的气相色谱分析条件下,以Y代表组分的峰面积,以X代表组分的质量
(μg),DBP的线性回归方程为Y=327.47X+0.52,相关系数为0.9998;DEHP的回归方程为Y = 485.43 X +3.71,相关系数为0.9997。重复测定混合标准溶液6次以检验方法的精密度,DBP和DEHP的RSD分别为2.4%和2.9%。对试样分别进行加标回收试验,DBP和DEHP的回收率分别为90.6%和99.4%。以“1.5”节的气相色谱条件测定同批塑料食品袋,DBP和DEHP的含量分别为0.05%和0.12%。
2.8 方法比较
比较薄层扫描与气相色谱的分析结果,实验数据接近。以气相色谱测定结果为真实值,对薄层色谱测定方法进行t检验,当置信度为95%时,薄层色谱测定结果│t│< t 0.95 ,说明薄层色谱方法准确可靠。由于薄层色谱方法简便易行、样品用量少、前处理简单、分析时间短、样品花费低、灵敏度高,故可作为酞酸酯的定量检测方法。
参考文献:
[1] G6mez-Hens A,Aguilar-Caballos M P. TRAC-Trends in Analytical Chemistry,2003,22 (11):847
[2] Li xiujnan,Zeng Zhaorui,Chen Yin,Xu Ying. Talanta,2004,63:1013
[3] Chen Huiming,Wang Chao,Wang Xing.Chinese Journal of Chromatography (陈会明,王超,王星. 色谱),2004,22 (3):224
[4] Kambia K,Dine T,Gressier B,Germe A F,Luyckx M,Bmnet C,Michaud L,Gottrand F. J Chromatogr B,2001,755:297
[5] Shen Haoyu. Talanta,2005,66 (3):734
[6] Lin Xingtao,Wang Xiaoyi,Ren Ren Environmental Poilution and Control (林兴桃,王小逸,任仁. 环境污染与防治),2003,25 (5):286
[7] do Nascimento Filho I,von Muhlen C,Schossler P,Bastos Caramao E. Chemosphere,2003,50(5):657
[8] Du Bing,Zhang Pengyi,Zhang Zulin,Yu Gang. Environmental Science (杜兵,张彭义,张祖麟,余刚. 环境科学),2004,25 (1):114
[9] Cai Yaqi,Jiang Guibin,Liu Jingfu. Anal Sci,2003,l9 (11):l491
[10] Toda H,Sako K,Yagome Y,Nakamura T. Anal Chim Acta.2004.519:213