-
+关注
私聊
-

yhl-87_
第21楼2009/11/06
薄层色谱法应用系列讲座(18)-测定刺五加注射液中紫丁香苷
薄层色谱扫描法测定刺五加注射液中紫丁香苷的含量
摘要
目的:采用薄层色谱扫描测定刺五加注射液中紫丁香苷的含量。
方法:采用日本岛津CS - 930型扫描仪,检测波长为266 nm。
结果: 紫丁香苷点样量在0.06μg~11μg的范围内与峰面积呈良好的线性关系,r为0.9984,回收率为105.8%。
结论:本方法操作简便、快速,结果准确可靠。
本文摘自2006年《药物分析杂志》,作者是山西大学化学化工学院的冯彦琳,李美萍,李俊,张生万等老师们,他们用薄层色谱扫描法定量分析刺五加中主要活性成分紫丁香苷,结果满意。现将全文介绍如下:
前言
刺五加注射液是由五加科植物刺五加提取加工精制而成的灭菌水溶液,具有平补肝肾、益精壮骨的作用。临床主要用于治疗各种心脑血管疾病。刺五加的主要活性成分为刺五加苷A、B、B1、C、D、E、F、G等,其中以刺五加苷B(紫丁香苷又名紫丁香甙)活性成分为最高,药理作用明显,刺五加类药物的质量标准通常以紫丁香苷的含量来确定。近年来,薄层色谱作为一种方便、快捷的分离手段已广泛应用于各类中草药指标性或有效成分的含量测定[1,2]。国内已相继报道过高效液相色谱法测定刺五加注射液中紫丁香苷的含量[3]、刺五加总甙提取物中刺五加甙B(丁香甙)薄层扫描测定[4]。前者操作费时且费用高,后者需二次展开,分离效果欠佳。对于薄层色谱扫描法测定刺五加注射液中紫丁香苷的含量未见报道。本文探讨并建立了对市售刺五加注射液中紫丁香苷含量进行薄层色谱扫描测定的新方法,该法操作简便快速、分离效果好,且结果令人满意,为进一步完善该制剂的质控标准提供了实验依据。
实验
1 仪器与试药
仪器:岛津CS - 930型薄层色谱扫描仪;定量毛细管(美国DRUMMOND SCIENTIFIC COMPA-NY);硅胶板GF254(青岛海洋化工厂分厂)。
试剂:丁香苷对照品(中国药品生物制品检定所);刺五加注射液(黑龙江省完达山制药厂;ZZ-5397 黑卫药准字(1983)第200038号);其他试剂均为分析纯。
2 溶液的制备
2·1 对照品溶液 精密称取紫丁香苷对照品适量,用甲醇配制成0.02 mg·mLˉ1和1 mg·mLˉ1的溶液,作为紫丁香苷对照品溶液。
2·2 供试品溶液 取本品直接作为测定紫丁香苷用供试品溶液。
3 展开体系及扫描波长的选择
3·1 展开体系的选择 曾用乙酸乙酯、甲醇、氯仿、丙酮、乙醇等十余种单一或混合展开剂,对标样和试样进行薄层分离试验,结果表明以乙酸乙酯一氯仿一甲醇(1: 0.8: 0.8)作展开剂,分离效果好(如图1)。
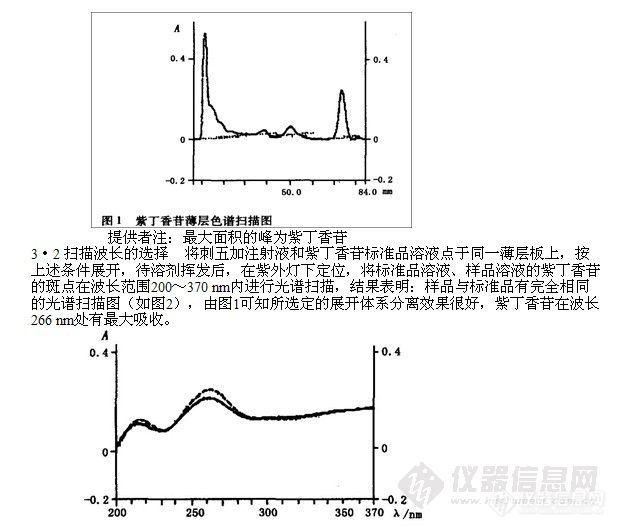
4 稳定性实验
按实验方法将试样展开并每隔10 min进行1次色谱扫描测定,取4 h内的测定结果,以峰面积A对时间t作图得图3,从图3可知,峰面积值变化不大,待测组分的测定值在4 h内稳定。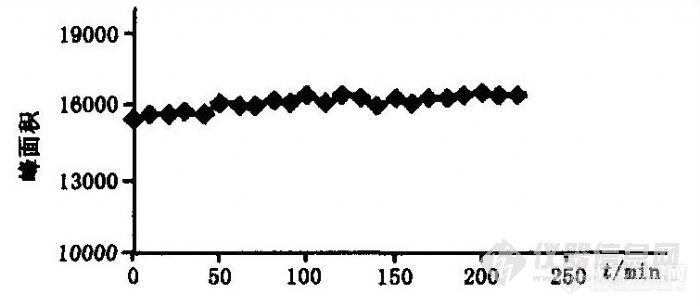
5 线性范围
将不同量的标准品溶液分别点于同一薄层板上,按上述实验条件展开,进行薄层色谱扫描测定。以点样量对峰面积作图,结果表明点样量在0.06μg~11μg范围内呈线性关系。回归方程为: y=30771X+10482 r =0.9984
6 精密度测定′
在同一块薄层板上点相同量标准品5个点,按上述实验条件展开后,用薄层色谱扫描仪依顺序测定5个斑点的峰面积。平均面积值为245185,RSD为0.48%。
7 样品测定
准确吸取刺五加注射液1 μL,浓度为0.02 mg·mLˉ1和1 mg·mLˉ1的标准品溶液各3 μL,交叉点于同一板上,按上述实验条件展开后进行薄层色谱扫描外标二点法定量测定紫丁香苷含量。4次测定结果分别为0.334,0.397,0.369,0.379μg·μLˉ1,平均含量为0.378μg·μLˉ1。
8 回收率测定
准确吸取注射液1μL,浓度为0.02 mg·mLˉ1和1 mg·mLˉ1的标准品溶液各3μL,交叉点于同一板上,然后在样品斑点上各点1 mg·mLˉ1的标准品溶液各1μL,按上述实验方法展开测定,回收率分别为104.9%, 105.6%, 105.8%, 105.6%; RSD 为0.37%。
9 讨论
9.1 本定量技术关键是选展开剂,使被测组分与其他组分分离,才能更好地进行实验。
9·2 本文所研究的刺五加注射液中紫丁香苷含量测定方法可为完善该制剂的质控标准提供实验依据。
-
+关注
私聊
-
yhl-87_
第22楼2009/11/06
薄层色谱法应用系列讲座(19)-小儿消积丸含量检测
摘 要
目的:研究小儿消积丸定性定量检测方法。
方法:采用TLC法对处方中的大黄、香附、牵牛子、槟榔4种药材进行定性鉴别;采用HPLC法对处方中大黄的大黄素和大黄酚进行了含量测定。
条件:保护柱:Symmetry-C18(4.6 mm×20mm×5μm);分析柱:Kromasil-C18(4.6 nim×150 mm×5μm);流动相:甲醇-0.1%磷酸溶液(85:15);流速:1 mL·minˉ1;检测波长:254 nm;柱温:室温。
结果:大黄素在2.06~65.92μg·mLˉ1范围内线性关系良好,r=0.9995,平均回收率为98.6%,RSD为1.7%;大黄酚在2.14~68.48μg·mLˉ1∶范围内线性关系良好,r=0.9993,平均回收率为98.l%,RSD为1.4%。
结论:本方法准确、可靠地进行定性、定量检测,能有效控制小儿消积丸的质量。
本文由山东省聊城市药品检验所的王登旭和杨瑞恩及中国药品生物制品检定所的魏玲等研究人员用薄层色谱分析小儿中成药的成分,用HPLC分析主要成分的含量。由于薄层板原图背景和斑点均带彩色,而黑白拍照印刷效果稍差点。现全文介绍如下:
前 言
小儿消积丸质量标准现载于《中华人民共和国卫生部药品标准》中药成方制剂第二册,是由大黄、木香、陈皮、黄芩、牵牛子、香附、三棱、槟榔等15味中药制成的水丸,主要用于小儿消食积滞、脘腹涨痛、面色萎黄、身体瘦弱等,多年临床验证,疗效显著。原标准只有丸剂项下的检查项目,不能反映其质量,经检索,国内外未见对小儿消积丸定性定量检测方法研究的相关报道。为有效控制产品质量,本文建立TLC法对大黄、牵牛子、香附、槟榔进行定性鉴别,用HPLC法对大黄中的主要成分大黄素和大黄酚进行含量测定。结果表明方法灵敏、准确、重复性好,可用于本品的质量控制。
实验
1 仪器和试剂
美国TSP - P200 HPLC色谱仪、UV100检测器;美国Anastar工作站;层析缸、薄层板涂布器、硅胶G和硅胶H(青岛海洋化工厂);超声波药品处理器(山东济宁超声波仪器厂)。大黄酸对照品、α-香附酮对照品、牵牛子对照药材、槟榔对照药材(中国药品生物制品检定所提供,鉴别用),大黄素、大黄酚对照品(中国药品生物制品检定所提供,含量测定用);甲醇为色谱纯,水为超纯水,其他试剂均为分析纯。小儿消积丸及阴性样品(东阿阿胶集团临清华威药业有限责任公司)。
2 方法与结果
2.1 薄层色谱鉴别
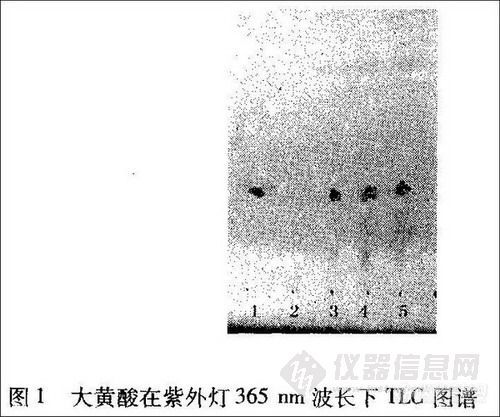
2.1.1 大黄 取本品0.3 g,研细,依法[l]操作,展开8 cm,取出,晾干,置紫外光灯(365 nm)下检视。色谱中供试品(4、5号斑点)与对照品(1、3号斑点)在色谱相应的位置上,显颜色相同的荧光斑点,Rf值为0.42,阴性样品(2号斑点)则无斑点。结果见图1
香附 取本品粉末3 g,依法[2]操作,展开8cm,取出,晾干,置紫外光灯(254 nm)下检视。色谱中供试品(5号)与对照品(1、2、3号)均在色谱相应的位置上,显相同的深蓝色斑点,Rf值为0.80。阴性样品(4号)则无斑点。结果见图2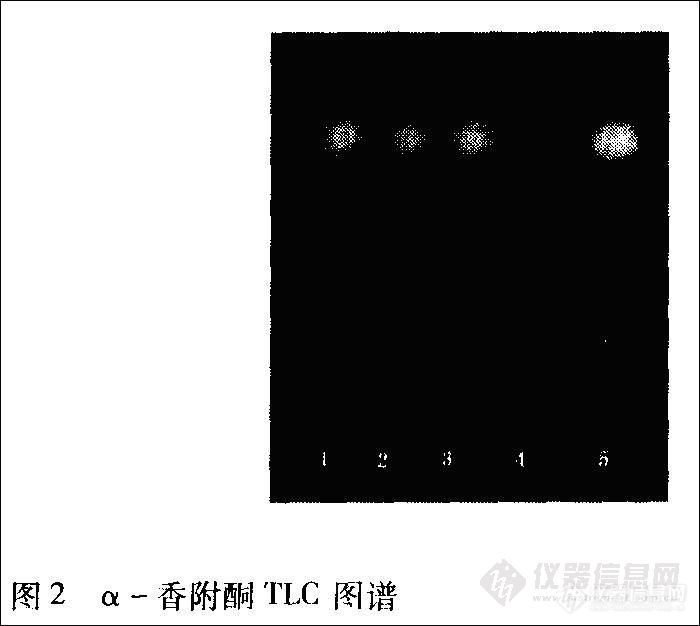
2·1·3 牵牛子 取本品3 g,研细,依法[3]操作,展开9 cm,取出,晾干,喷以5%香草醛溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,Rf值为0.71。阴性样品则无斑点。结果见图3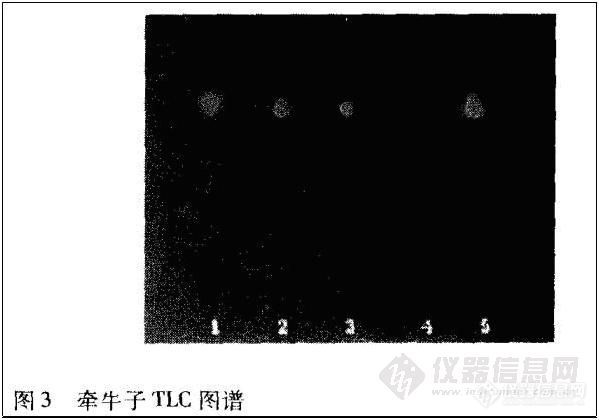
2·1·4 槟榔 取本品粉末10 g,依法[4]操作,展开9 cm,取出,热风吹干,喷以稀碘化铋钾试液。色谱中供试品(1、2、3号)在与对照品(5号)色谱相应的位置上,显相同颜色的斑点,Rf值为0.49。阴性样品(4号)则无斑点。结果见图4。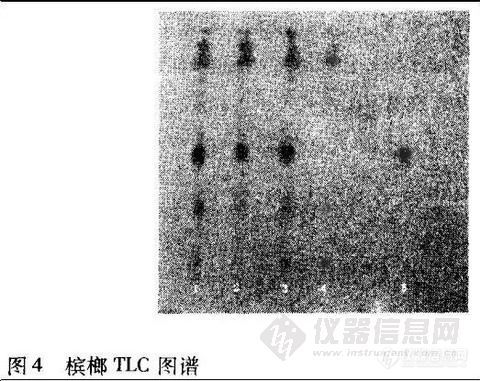
2·2 含量测定
2.2.1 HPLC色谱条件
保护柱:Symmetry -C18(4.6 mm×20mm×5μm);分析柱:Kromasil-C18(4.6 nim×150 mm×5μm);流动相:甲醇-0.1%磷酸溶液(85:15);流速:1 mL·minˉ1;检测波长:254 nm;柱温:室温;进样量:l0μL。在上述色谱条件下,大黄素和大黄酚的保留时间分别为7.0 min和9.1 min;理论板数以大黄素计为9500;以大黄酚计为10000;大黄素和大黄酚的分离度大于2.0。
2.2.2 线性关系考察
精密称取大黄素、大黄酚适量,用甲醇分别稀释成大黄素浓度为2.06,4.12,8.24,16.48,32.96,65.92μg·mLˉ1的对照品溶液以及大黄酚浓度为2.14,4.28,8.56,17.12,34.24,68.48 1μg·mLˉ1的对照品溶液。在上述色谱条件下,分别进样l0μL,记录色谱图,以峰面积对进样浓度进行线性回归(n=6),大黄素和大黄酚回归方程和相关系数分别为:
y=755X-5.62l r=0.9995
y=642X-I9.81 r=0.9993
结果表明大黄素浓度在2.06~65.92μg·mLˉ1范围内、大黄酚浓度在2.14~68.48 lμg·mLˉ1范围内与峰面积线性关系良好。
2.2.3 精密度试验
精密吸取8.24μg·mLˉ1的大黄素对照品溶液和17.12μg·mLˉ1的大黄酚对照品溶液各l0μL,分别进样5次,大黄素峰面积的RSD为1.1%,大黄酚峰面积的RSD为1.3%。表明精密度良好。
2.2.4 重复性试验
取同一批号(20030301)样品6份,按“2.2.8”项下的方法处理、测定,大黄素含量的HSD为0.6%,大黄酚含量的R5D为0.9%。
2·2·5 稳定性试验
精密吸取供试品溶液(批号:2003030 I),在0,l,2,4,8,12,24 h测定,大黄素峰面积的RSD为1.3%,大黄酚峰面积的RSD为1.3%。表明供试品溶液稳定性良好。
2.2.6 加样回收率试验
精密称取已知大黄素、大黄酚含量的样品
-
+关注
私聊
-
yhl-87_
第23楼2009/11/06
薄层色谱法应用系列讲座(20)-测定烟草中β-胡萝卜素
高效薄层色谱扫描法测定烟草中β-胡萝卜素
摘要 建立了测定烟草中β-胡萝卜素的高效薄层色谱扫描方法,以V(苯):V(丙酮) = 4:1为展开剂,检测波长450nm,线性范围50ng~0.5μg,最低检测限20ng。
本文由合肥经济技术学院的时亮、朱晓兰、刘百战和高芸老师们共同完成,现具体介绍如下:
1 前言
胡萝卜素类物质是烟草中许多致香物质的前体。据报道,它们的降解产物与烟草的香味有着密切的联系⑴~⑶。在烟草的各类胡萝卜素中,β-胡萝卜素的作用十分重要。因此研究和测定烟草及烟制品中β-胡萝卜素的质量比对于研究烟草配方有着更重要的意义。本文用高效薄层色谱法测定了烟草中β-胡萝卜素的质量比,与其它方法相比,它具有快速、灵敏、简便等特点,测定结果令人满意。
2 实验部分
2.1实验仪器与试剂
岛津CS-9000双波长薄层扫描仪,ZFQ85A旋转蒸发仪,规格为200mm×100mm的HSGF254的高效薄层硅胶板 (烟台化工研究所制),于105℃下活化1 h,置于干燥器中备用。
苯、丙酮皆为分析纯,β-胡萝卜素纯度大于99.9%。以丙酮为溶剂,配成50mg/L的β-胡萝卜素标准溶液,置于冷暗处备用。
2.2实验方法
用微量定量点样管将β-胡萝卜素标准溶液和样品溶液点于上述经活化后的HSGF254高效薄层硅胶板上。以V(苯):V(丙酮)=4:1为展开剂,上行展开至15cm。取出硅胶板,将其置于薄层扫描仪上,以双波长反射法锯齿型扫描。测定波长为450nm,参比波长为520nm,散射系数SX=3。根据峰面积积分值,以外标法定量。
3 结果与讨论
3.1 分离条件及测定波长的选择
曾用乙酸乙醇、甲醇、乙醇、丙酮、苯、石油醚等单一或混合展开剂,对标样及试样进行薄层分离试验,结果表明以V(苯):V(丙酮)= 4:1混合展开剂分离效果为最佳。对展开后的β-胡萝卜素斑点进行原位可见光谱扫描可知,在450nm处β-胡萝卜素有最大吸收。因此,选择450nm为检测波长,选择吸收值较低的520nm为参比波长。
3.2 线性关系的考察
精确量取已配制好的标准溶液1、2、3、4、5μL点样于同一硅胶板上,按上述实验条件展开、晾干、扫描,以峰面积积分值Y为纵坐标,标准品质量X(μg)为横坐标,计算其回归方程为Y= 466102X-16150,r =0.9989。β-胡萝卜素在50ng~0.5μg范围内呈线性关系,最低检测限20ng。
3.3 烟草样品测定
分别准确称取烟草烘烤过程不同阶段的烟叶2g,与V(3OmL,丙酮):V(水)= 9:1一起放入旋转研磨器中磨成碎末。离心后,取上层清液于分液漏斗,加入适量苯,再缓慢加入水。反复萃取,使色素转移到苯层中。经无水硫酸钠脱水后,用旋转蒸发仪在40℃下减压浓缩至近干,残留物用苯溶解并定容至5mI,。再按实验方法点样、展开,扫描。测定结果见表-1。
表-1 烟草试样测定结果(n=3) 7474-1

3.4 精密度
以相同体积的β-胡萝卜素标准溶液5份,在同一薄层板上点样、展开、扫描,进行同板精密度测定,RSD为2.02%。同法进行异板精密度测定,RSD为3.25%。
3.5 稳定性
精确量取不同体积的β-胡萝卜素标准溶液在同一薄层板上点样、展开,每隔20min扫描测定一次。结果表明,90min内斑点面积基本稳定,RSD为2.56%。
3.6 回收率
将样品和加入已知量β-胡萝卜素的样品分别点样、展开及扫描测定,用差减法求出β-胡萝卜素的回收率,结果见表-2。
表-2 回收率测定结果(n=5) 7415-1
Table2 Recovery ofβ-carotene added to sample (n=5)
参考文献
1、 Chortyk O T.Tobacco Science,1967,11:137—139
2、 Severson R F,Arrendale R F,Chaplin J F et a1.J Agric Food Chem,1979,27(4):896~898
3、 周冀衡.烟草生理与生物化学.合肥;中国科学技术大学出版社,1996.401~402
-
+关注
私聊
-
yhl-87_
第24楼2009/11/06
薄层色谱法应用系列讲座(21)-安中片的TLC/HPLC定量
用TLC与HPLC方法对安中片的主要成分
质量控制标准研究
摘 要 目的:建立安中片的质量控制方法。
方法:采用TLC方法对安中片中的延胡索、甘草进行定性鉴别,采用HPLC法,All-tima C18色谱柱(250 mm×4.6 mm×5 um),以甲醇-1%醋酸(45:55)为流动相,检测波长285 nm,柱温40℃,流速1 mL·minˉ1,进样量10μL,对安中片中的肉桂酸进行了含量测定。
结果:延胡索、甘草的薄层色谱鉴别专属性强,肉桂酸进样量在0.004~0.104μg范围内与峰面积呈良好的线性关系(r=0.9999,n=8)。肉桂酸的平均回收率为98.6%,RSD=1.6%。
结论:本方法可准确地进行定性、定量,可用于控制安中片的质量。
本文由邓少伟1,程显隆2,马双成2 (l专家中药品种保护审评委员会;2中国药品生物制品检定所)多位研究人员用TLC、HPLC方法完成安中片主要成分定量分析,现全文介绍如下
前 言
安中片收载了中华人民共和国卫生部药品标准中药成方制剂第五册[1],由桂枝、延胡索、牡蛎、小茴香、砂仁、高良姜和甘草共7味药材组成。具温中散寒、理气止痛、和胃止呕之功效。用于胃脘疼痛、慢性胃炎、胃酸过多、胃及十二指肠溃疡。原标准中没有薄层鉴别和含量测定项目,为了控制该制剂的质量,保证临床用药安全、有效,特选择方中的主要药味作为质量控制指标,进行分析研究。本研究通过实验建立了方中的主要药味延胡索和甘草2个薄层鉴别项目,其余药味在薄层鉴别色谱中有不同程度的干扰。桂枝为该制剂的君药,其主要成分为肉桂酸。肉桂酸的H PLC法含量测定已有文献报道[2~4],但不适于该制剂的含量分析,通过实验并参考文献[2~4],采用HPLC法测定了制剂中肉桂酸的含量,提高了质量控制标准,可用于该药的质量控制。
实 验
1 仪器与试药
高效液相色谱仪:Shimadzu液相色谱系统,包括LC -10AT泵、SPD -10A检测器、SIL -10AD进样器、DGU -14A脱气装置、CTO-10A柱温箱。
试验用样品(安中片及阴性样品)均由中国药品生物制品检定所中药室按处方自制,批号分别为:030201,030202,030203。含量测定用对照品延胡索乙素、肉桂酸以及对照药材延胡索和甘草均由中国药品生物制品检定所提供。薄层硅胶预制板(青岛海洋化工厂)。甲醇为色谱纯,乙醚、正己烷、氯仿、甲醇、冰醋酸等均为分析纯,纯净水为Milli-Q纯水。
2 薄层鉴别
2.1 延胡索的鉴别
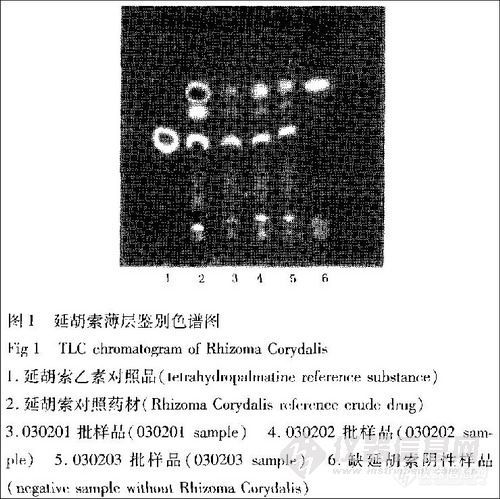
取本品6片,研细,滴加氨水,使润湿,加乙醚30 mL超声(功率250 W,频率40kHz)处理20min,滤过,滤液浓缩至干,加甲醇1 mL使溶解,作为供试品溶液;取缺延胡索的阴性样品,依法制成阴性对照溶液;另取延胡索乙素对照品,加甲醇制成每1 mL含0.5 mg的溶液,作为对照品溶液;再取延胡索对照药材0.5g,同法制成对照药材溶液。照薄层色谱法(中国药典2005版一部附录ⅥB)试验,吸取上述4种溶液各5 μL,分别点于同一硅胶G薄层板上,以正己烷一氯仿一甲醇(l0∶5∶1)为展开剂,于氨饱和蒸气中展开,展开8cm,取出,晾干,碘熏至斑点显色清晰,置紫外灯(365 nm)下检视。结果样品色谱中,在与对照品及对照药材色谱相应的位置上,显相同颜色的荧光斑点,而阴性对照无干扰(图1)。
2.2 甘草的鉴别
取本品6片,研细,加甲醇15mL,超声(功率250W,频率40 kHz)处理20 min,滤过,滤液浓缩至干,加甲醇l mL使溶解,作为供试品溶液;取缺甘草的阴性样品,依法制成阴性对照溶液;另取甘草对照药材0.1g,同法制成对照药材溶液。照薄层色谱法(中国药典2005年版一部附录ⅥB)试验,吸取上述3种溶液各5μL,分别点于同一硅胶G薄层板上,以氯仿一甲醇(8:1)为展开剂,展开8 cm,取出,晾干,置紫外灯(365 nm)下检视。结果样品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,而阴性对照无干扰(图2)。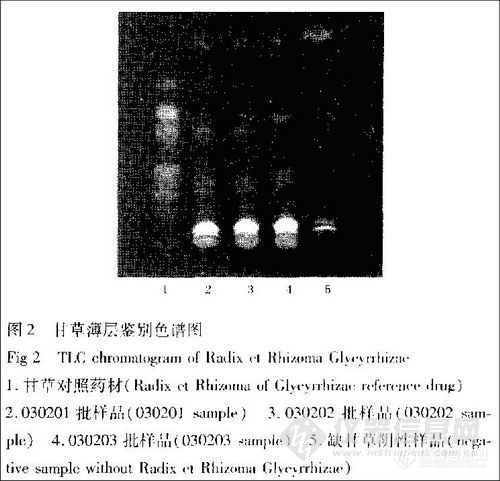
3 含量测定
3.1 溶液制备
3.1.1 对照品溶液 精密称取肉柱酸对照品适量,加甲醇制成每I mL中含 4 μg 的溶液,即得。
3.1.2 供试品溶液 取本品10片.除去糖依.精密称定,研细,再取粉末约1 g,精密称定,置具塞锥形瓶中,加甲醇25 mL.密塞。称定质量.超声(功率250W.频率40 kHz)处理30min,放冷.再称定质量,用甲醇补足减失的质量.摇匀,取上清液过0 45 μm的微孔滤膜,即得。
3.1.3 阴性对照溶液 取缺桂枝的阴性样品同“3,1.2”项下方法操作,即得。
3.2 色谱条件与系统适应性实验
色谱柱:Alltima C18,.色谱柱(250 mm×4.6 mm,5 μm),流动相:甲醇-l%醋酸(45:55)。检测波长:285nm ,40 ℃,流速:1 mL·minˉ1 .进样量:10μL。在此色谱条件下.肉桂酸对照品、安中片和缺桂枝阴性样品的分离色谱图见图3。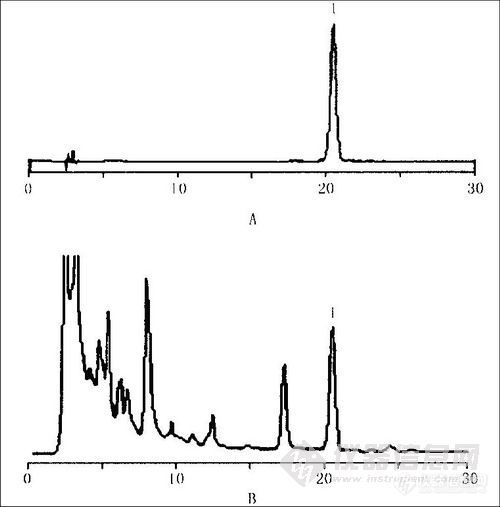
3.3 线性关系考察
按上述色谱条件,分别精密吸取上述对照品溶液I,2,4,8,10,l5,20,30μL注入液相色谱仪.测得峰面积。以峰面积值为纵座标,内佳酸对照品进样量(μg)为横座标.绘制标准计算回归方程:
y=8.2×106 X -4.6×103 r=0.9999
在0.004~0.104μg 范围内,肉桂酸对照品进样量与峰面积呈良好的线性关系。
3.4 精密度试验
精密吸取上述对照品溶液10μL,.按上述色谱条件重复进样5次.测定峰面积.RSD为0.6%。
3.5 稳定性试验
精密吸取上述对照品溶液10μL.按上述色谱条件分别于0,3,6,12,13,l7,21.28h测定峰面积,计算RSD为2.2%。结果表明对照品在28 h 内稳定。
3.6 重复性试验
取样品 50片、除去糖衣.精密称定,研细,混和均匀。精密称取6份,每份约l g 按¨3. I. 2”项下方法处理得供试品溶液,依上述色谱条件测定含量,RSD为 0.7%。
3.7 回收率试验
精密称取“3,6”项下的细粉5份,每份约0.5g,精密称定。再精密加入肉桂酸对照品甲醇溶液(0.00208 mg·mLˉ1)25 mL,按“3,I,2”项下方法处理得所需溶液,再按上述色谱条件测定含量,计算平均回收率为98.6%,RSD为1.6%。
3.8 样品测定
精密吸取供试品溶液及对照品溶液,依上述色谱条件测定,每批样品测定2份,以外标法计算含量,取其含量平均值。结果见表l。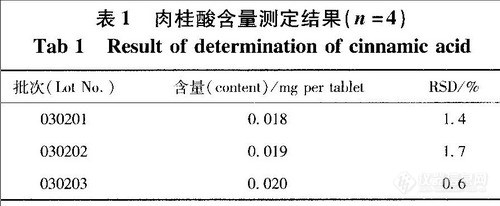
4 讨论
本文对安中片进行了质量控制标准研究,增加了延胡索和甘草的薄层鉴别,建立了采用高效液相色谱法测定处方中桂枝的有效成分肉桂酸含量的方法。样品处理方法简单,薄层色谱图清晰,含量测定结果准确,重复性良好,可用于该制剂的质量控制。
-
+关注
私聊
-
yhl-87_
第26楼2009/11/06
薄层色谱法应用系列讲座-TLC-2MS联用技术进展(22)
薄层色谱-双质谱联用技术及其进展
摘 要: 概述了薄层色谱同质谱的联用( TLC2MS) 技术及其进展,着重介绍了TLC-2MS 联用技术中的多种接口及已经开始广泛使用的几种较先进的电离方法。
本文由北京化工大学理学院的董慧茹, 张建军研究人员对薄层色谱双质谱联用技术中的各种接口、电离方法等现状及进展做了评述,现全文介绍如下。
前 言
近年来,随着新化合物的迅速增加,样品的复杂程度越来越高。复杂体系的分离和测定已成为分析工作者所面临的艰巨任务。由液相色谱、气相色谱、超临界流体色谱和毛细管电泳等组成的色谱学可以解决现代分离、分析的许多问题,因此正在飞速发展,以色谱和光谱技术为基础所发展的各种联用技术已成为当今分析化学研究的热点之一。
1990 年2 月在比利时召开的第一届国际色谱联用技术会议上,有关气相色谱、高效液相色谱、毛细管电泳等分离技术与傅立叶变换红外、质谱、原子光谱的联用均有报道[1 ] 。在众多的联用技术中,薄层色谱与质谱的联用出现得较晚,有关报道也较少。
实 验
1 薄层色谱-双质谱的联用及偶联方式
薄层色谱( TLC) 是一种分离效率高、成本低、样品用量少、应用广泛的微量分离手段;而质谱(MS)是一种灵敏度高、选择性好、可进行有效定性分析的现代仪器。因此两者的联用,实现了优势互补,为复杂样品的定性提供了一条有效的途径。
1. 1 TLC-2MS 联用的主要问题
实现TLC 与MS 联用对薄层色谱来说,总希望待测混合物能真实并毫无损失在薄层板上有效分离,每一个斑点只代表一种纯净的组分,以便于设计同质谱联用的接口。但是,由于分离后的薄层色谱体系包括过量吸附剂、粘和剂、荧光指示剂、残余溶剂和盐等,每一个斑点都很难达到完全纯净,因而对接口提出了更高的要求。要么,接口能够除去过量的吸附剂,使待测组分从吸附剂上有效解吸,并浓缩精制;要么,质谱对待测组分有高的选择性,在检测色谱体系时,仅对待测组分有高的分辨率,不受其它无关物质的影响。这样,就需要优化样品注入体系。
其次,实现TLC 与MS 联用,还要考虑质谱中样品分子的电离方法。使分子电离的方法很多,传统的电子轰击源( EI) 和化学电离源(CI) 均可应用于TLC-2MS。此外,还有一些新的分子离子化方法,诸如快原子轰击( FAB),二次离子质谱(SIMS) ,基质辅助激光解吸(MALD) 等。这些新的离子源的使用使TLC2MS 在化合物定性方面更加快速、准确、灵活,大大扩展了薄层色谱的应用。
1. 2 TLC-2MS 的偶联方式
TLC-2MS 的偶联方式关系到从分离到鉴定过程中待测组分的转移。可分为下列三种:
(1) 待测化合物从吸附剂中洗脱提取,精制收集,然后作为纯组分进入质谱,称之为“手动方式”。在这种方式下,薄层色谱只是一种提纯工具。
(2) 样品不必同吸附剂分离,将样品斑点刮下后,同吸附剂粉末一起引入质谱电离室,或将一小块薄层板切除坯连同斑点一起引入质谱电离室。
(3) 整个色谱体系放置于质谱的电离室并直接进行分析。该方法难度较大,需要对质谱计进行大的改进,但可以提供理想的质谱数据。后两者称为“仪器方式”。
2 TLC-2MS 的接口技术
要建立薄层色谱同质谱的偶联,首先要解决两者之间的接口问题。接口的方式多种多样,一个优秀的接口由被分析物质的特性和所采用的电离方法而定。
2. 1 提纯直接引入法
这是实现TLC2MS 联用的最简单方法,也是广泛采用的方法。在这种方法中, TLC 和MS 的联用是离线的,薄层色谱仅作为一种分离提纯方式。样品经薄层色谱分离后,斑点从板上移除,用适当的溶剂洗脱,蒸去溶剂后精制得纯品,再将纯品注入质谱计进行分析。
苟丽萍等[2 ]用所述方法分离并鉴定了4 ,4′2二氨基二苯砜,确定了其分子结构。Henion 等[3 ] 用TLC-2MS 离线分析鉴定了多种用于治疗马类疾病的药物。Queckenbery 等[4 ]使用包括TLC-2MS 在内的多种联用技术,分析了从一种野生植物中提取出来的复杂酰胺类化合物,使用了包括化学衍生、UV、FTIR 在内的检测方法,并对各种方法进行了比较。该方法的不足之处是组分在转移过程中易损失,易氧化,容易被污染,操作周期长等。
2. 2 热蒸发法
该方法适用于易挥发、热稳定、分子量小的样品,样品分子受热蒸发从而直接同吸附剂分离。
Parkhurst 等[5 ] 将薄层板置于一试管中,周围绕上加热线圈,可对薄层板进行选择性加热。或者将薄层板放置在靠近质谱计的平台上,平台可移动,用固定强度的光源或激光对薄层板分离区域依次加热,再用气流将蒸发解吸的样品吹入质谱计。XiaoChen 等[6 ]将斑点从薄层板上刮下直接置于试样池中进行加热萃取,并以He 作载气,GC-2MS 测定了七种萘化物、四种环芳烃(PAH) 及三种杀虫剂。
热蒸发法方便迅速,连续性好,是一种比较好的TLC-2MS 接口。如果色谱基质不挥发而样品是挥发的,可以将样品和色谱基质一起放在质谱的直接进样探头中,当探头受热时,样品挥发,从而可以用传统的EI 源或CI 源将样品分子电离而得到质谱图。Hutzinger 等[7 ,8 ]曾用这种技术鉴定了吲哚类物质。Kraft 等[9 ]用该法分析了酚类、甾族化合物、核苷及氨基酸类混合物
2. 3 特殊洗脱技术
特殊的洗脱技术包括许多设计巧妙的仪器装置和方法,使溶剂能有效将样品洗脱并迅速转移至质谱计,连续性好。该技术的一种较简单的TLC2MS接口就是将移除的吸附剂和样品同时装柱,溶剂从上往下对样品进行洗脱,并通过一个玻璃毛细管直接引入质谱计的电离室,适用于直接进行FAB 分析的一种接口是将样品经薄层色谱分离后,将一种具有双层胶带(double2stick tape) 的探头按在斑点上,将待分析的吸附剂斑点粘贴,再将合适的溶剂加到粘在探头顶端的吸附剂上,充分提取后,直接进入FAB 源分析[10 ] 。
Somoyi 等[11 ]使用改进的灯芯2量杆技术进行了TLC-2MS 分析。斑点定位后从薄层板上刮除,样品和吸附剂放入浓缩装置中,将氯化铵三棱体浸入容器底部的溶剂中,当溶剂从三棱体顶端挥发出来的时候,样品也从吸附剂中洗脱出来。当溶剂蒸发完全后,切除三棱体顶端,碾碎,将样品和氯化铵粉末一同由直接进样探头送入质谱检测。
Hisao 等[12 ]使用了一种特殊的洗脱技术,即利用“梯形”对样品进行浓缩,使样品斑点浓缩成一条直线,再用FAB 质谱检测。他们已用这种技术测定了酸红、霉菌素、胱氨酸等物质在正相和反相薄层板上的Rf 值,并作出其FAB 质谱图。
2. 4 毛细管技术
毛细管可以广泛用于TLC-2MS 接口上。Brown等[13 ]设计了一种装置,该装置利用高温高压迫使溶剂从薄层板切除坯中渗滤从而移除样品,快速流过转移毛细管,直接进入FAB 或其他电离室,这与加压薄层色谱原理相似。但不需要泵,因而该设备小巧易制,连续性好。
如果样品在吸附剂上吸附很强,可将薄层体系放在质谱计的真空室中。直接进样探头中装一内径50μm 的硅毛细管,毛细管接有注射泵。溶剂以225ml·min - 1的速度由硅毛细管流到斑点表面,斑点已在轰击焦点上, 电离后即可得样品的FAB 质谱图。
Brown 用上述技术成功分析了100ng 量的杀虫剂衍生物。由于可以方便调节FAB 底液的组成和极性,提高了洗脱效率和FAB 质谱图的分辨率。
Anderson 等[14 ]设计了一种TLC 和ESI 的偶联装置,称之为毛细管探头,该探头的工作原理同圆珠笔芯很相似。探头顶端的直径为0. 5mm ,可以很方便地同注射器、微型吸管及熔点毛细管等联接。
毛细管的内径小,在表面张力作用下利于转移微量液体,所以在TLC2MS 接口中应用很广。
-
+关注
私聊
-
yhl-87_
第27楼2009/11/06
薄层色谱法应用系列讲座-TLC-2MS联用技术进展(22) (下)
3 TLC2MS 中的电离方法
对质谱来说,和薄层色谱偶联的电离方法有很多种,除了传统的EI 和CI 源外,还有FAB、SIMS、MALD、ESI 等。Somen 等[15 ]对TLC 同多种现代光谱技术联用的进展进行了综述,着重介绍了同TLC联用的各种质谱电离方法,并介绍药物、抗生素、类脂、染料等在不同电离方式下的质谱图。现将各种电离技术与TLC 联用的情况作简要介绍。
3. 1 快原子轰击电离( FAB)
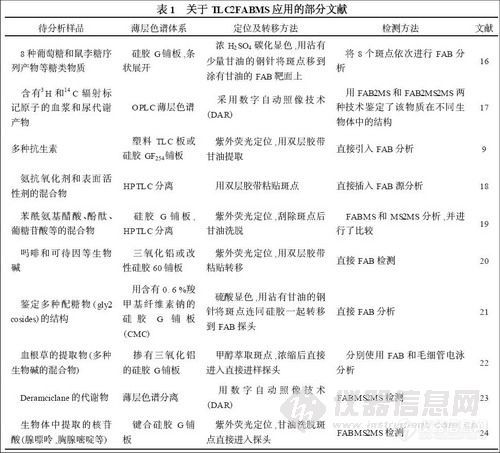
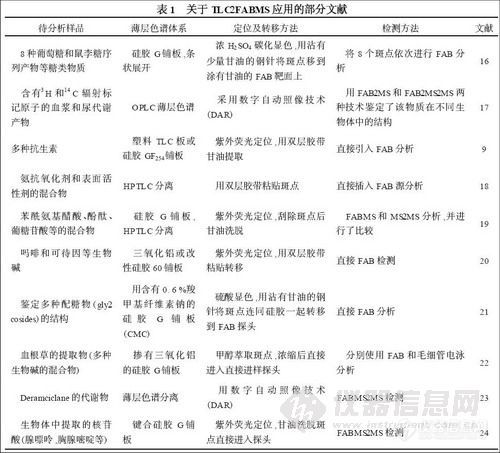
FAB 是质谱中广泛应用的电离方法之一。胡耀铭等[16 ]曾介绍过FAB 质谱与纸色谱和薄层色谱的联用,并举例说明了极性大,难挥发和热不稳定的多组分体系的分析方法。关于TLC-2FABMS 的报道还有很多,如表1 所示。
TLC-2FABMS 的缺点是只包含少量信息,仅能提供分子量和有限的碎片离子,存在背景和共色谱干扰等。因此,考虑用串联质谱来提高检测质量。
3. 2 二次离子质谱( SIMS)
SIMS 以其高灵敏度、宽的动态范围和优良的深度分辨,已逐步发展成为一种很有特色的表面分析手段,特别适于和TLC 联用,可详细提供薄层板上斑点的样品分布图象。
在TLC-2SIMS 中,一般需将离子从薄层斑点表面溅射出来,再由四极滤质器加速后,送入SIMS 源进行质谱分析。SIMS 包括液相SIMS (LSIMS) 和飞行时间SIMS( TOF SIMS)。
Masuda[25 ]等用浸有石蜡的氧化铝铺板,有效分析了酸红、叶红素和焰红,用三甘油定位浓缩后进行SIMS 分析, 0. 1μg 样品即可得到满意的质谱图。Kushi 等[26 ]用三已胺作洗脱剂, 吲哚或考马斯蓝定位, 用TLC2SIMS 分析了类脂。Unger 等[27 ] 用TLC 分离了蘑菇组织中的四元生物碱,不用洗脱溶剂,直接将蕈毒碱从纤维素板上溅射出来,得到了SIMS 质谱图。Yang 等[28 ]用TLC2SIMS 分析了有机锍盐的结构。
Nukagawa 等[29 ]用扫描TLC2LSIMS 分析了药物抗生素和其它有机化合物的混合物。Dunphy等[30 ]利用TLC2LSIMS 技术鉴定了胆汁酸盐的结构,并提供了该物质的正离子和负离子在薄层板上的三维图象。
TOF SIMS 用高速宽直径离子束从表面溅射二次离子,保留了样品在薄层表面的空间分布,可获得高分辨率的空间分布图。潘永艺等[31 ]用银载体分离并用TOF2SIMS 测得环氧树脂的质谱。Busch等[32 ]用TLC2TOF SIMS 测出有机锍盐、氢氧化物等多种试样的质谱图。
3. 3 基质辅助激光解吸电离(MALD)
MALD 是广泛用于在TLC 板上直接进行分析的一种电离方法,特别适用于极性大、体积大、难挥发的生物大分子,可用来分析蛋白质、多肽、聚核苷酸、低聚糖等。它不需要洗脱溶剂,高热能激光照射到斑点表面使样品分子溅射出来。Busch 等[32 ] 用C18键合硅胶G 铺板,微量注射器点样,用MALD2MS 分析了生物分子胆汁酸和盐、核苷和核苷酸。Kubis 等[33 ]用聚酰胺铺板,利用MALD-2MS 测得了糖、嘌呤、多环芳烃等许多有机化合物的正离子和负离子质谱,Ramley 等[34 ]用该技术测定了菲的质谱图,并说明了激光脉冲对质谱图的影响。
3. 4 电喷雾电离( ESI)
ESI 是新近出现的一种“软电离”质谱技术,已有人研究了它与TLC 的联用。ESI 适于大分子化合物分子量的测定,还可对极性大、难挥发、易分解的糖甙类化合物进行分析。孙维星等[35 ]用TLC 和ESI2MS 联用成功测定了溶液中非共价复合物的化学计量数和键合常数。Anderson 等[14 ] 用TLC 和ESIMS 联用测得了三羧基三苯膦盐的正离子质谱图,检测出薄层板上2pg 量的样品。
Yang 等[28 ]曾将ESI 和LSI 作了比较,发现两者进行质谱分析的样品浓度不同;比较了同一种锍盐的ESI 和LSI 谱图,发现LSI 质谱比ESI 质谱包括更多的碎片离子峰。
4 TLC-2MS-2MS 技术
串联质谱可提供比单一质谱更详细的信息,且不受背景离子的干扰,不受共色谱体系污染等,因而受到分析工作者的普遍重视。将串联质谱同薄层色谱联用同TLC-2MS 联用一样,可采用“手动方式”或“仪器方式”连接。Wilson 等[36 ] 对HPTLC-2MS-2MS的进展及应用进行了综述,指出该技术广泛用于合成化合物(如药物、增塑剂) ,天然产物(如甾体、糖脂) 的分析;并指出串联质谱与TLC 联用优于传统的单一质谱与TLC 联用。Wilson 等[37 ] 用TLC-2MS-2MS 联用技术测定了植物提取物中的激素。Martin 等[38 ]用HPTLC 和FABMS 以及串联MS 联用技术鉴定了人尿中提取的安替比林和三种代谢产物,不必同吸附剂分离就进行了直接分析。Mon2aghan 等[39 ] ,用TLC-2MS-2MS 分析了抗氧化剂O 及酚类抗氧化剂。Lafont 等[40 ]报道了昆虫提取物中蜕皮激素的TLC-2MS-2MS 分析。
5 结论
TLC-2MS 联用是一种较先进的分离分析技术,它方便直接、灵敏度高、检测迅速、适用范围广,对定性分析有很高的准确度。但是,它需要特殊的样品引入技术及先进的电离源,而具有特殊电离源的质谱计往往体积大,价格昂贵。因此,TLC-2MS 联用的普及受到了限制,发展和应用远不及GC-2MS。应该看到,TLC 具有GC 所不具备的优点,因此,将TLC和MS 联用代表了科学发展的趋势。在吸附

-
+关注
私聊
-
yhl-87_
第28楼2009/11/06
薄层色谱法应用系列讲座-白藜芦醇及其糖苷异构体含量(23)
薄层荧光扫描法测定虎杖中的白藜芦醇和白黎芦醇糖苷
摘 要 目的:建立虎杖中白藜芦醇及其糖苷异构体含量的测定方法。
方法:以甲醇提取目标组分,以聚酰胺薄膜为固定相,苯一甲醇一甲酸(l0:5:1)为展开剂层析分离,用荧光扫描法定量,λEx=306 nm,λEm=400 nm,线性参数SX=3。
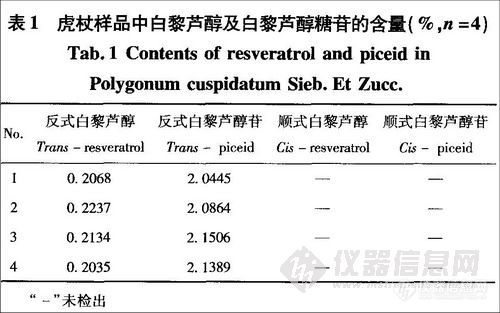
结果:4种组分(顺式白藜芦醇及其糖苷和反式白藜芦醇及其糖苷)的线性关系良好,相关系数为0.9942~0.9996,平均回收率为97.6%~98.6%,RSD为1.8%~3.1%。测定4个虎杖样品中反式白藜芦醇和反式白藜芦醇苷的含量分别为0.20%~0.22%和2.04%~2.14%,均未检出可定量测定的顺式异构体。
结论:方法简便、快速、灵敏、测定成本低。
前 言
常用中药虎杖(Polygonurn cuspidatum Sieb.Et Zucc.)具有祛风利湿,祛痰止咳,活血化淤等功效[I]。虎杖中含有白藜芦醇(resveratrol)及白藜芦醇苷(piceid)。研究发现,白藜芦醇和白藜芦醇苷具有抗肿瘤、抗凝血、抗高血脂症、抗菌、预防骨质疏松等多种生物活性[2,3]。天然白藜芦醇和白藜芦醇苷都存在反式(trans -)和顺式(cis -)二种构型,其中反式异构体的生物活性高于顺式异构体,反式的白藜芦醇或白藜芦醇苷在紫外线照射下均能转化为相应的顺式异构体[4]。
目前,虎杖中白藜芦醇及其糖苷异构体的测定主要用HPLC法[5],另外还有薄层层析一紫外分光光度法[6]、毛细管电泳法和化学发光法等[7,8]。 但这些方法多数只能测定游离白藜芦醇或白藜芦醇苷的总量,而不能分别测定它们的顺、反式异构体,因而测定结果不能反映虎杖中所含白藜芦醇及其糖苷异构体的准确情况。本文首次采用聚酰胺薄膜作固定相,利用荧光扫描法定量,不仅能同时测定4种异构体,且与硅胶作固定相的薄层层析法相比,测定的灵敏度提高了近100倍。方法简便、快速、灵敏、测定成本低。此法还可用于其他样品中白藜芦醇及其异构体的测定。
实 验
1 仪器与药品
CS -9301PC双波长飞点扫描分析仪(日本岛津);聚酰胺薄膜(8 cm×8 cm,浙江台州市路桥四甲生化塑料厂);微量进样器(10 μL,上海安亭微量进样器厂);ZF - C型三用紫外分析仪(254 nm,365nm,上海康禾光电仪器有限公司);LD - 02型高速中药粉碎机(浙江省温岭市大海药材器械厂)。
反式白藜芦醇对照品(纯度>99%,Sigma公司);反式向藜芦醇糖苷对照品(纯度>99%,复旦大学药学院天然药物研究室),苯、甲醇、甲酸、醋酸乙酯、氯仿、丙酮、醋酸、正己烷等均为分析纯;虎杖样品购自河南中医学院一附院。
2 溶液制备
2.1 对照品溶液
分别准确称取反式白藜芦醇和反式白藜芦醇苷对照品2.5 mg,用甲醇定容至25 mL,制成0.1 mg·mL-1的溶液。分别取以上两溶液适量,置365 nm紫外光下分别照射2 h和1 h,制得顺式白藜芦醇和顺式白藜芦醇苷对照品溶液,实验测得其转化率分别为69.3%和58.2%。
2.2 样品溶液
将虎杖粉碎后,准确称取虎杖粉约0.5g,用滤纸包裹后置于索氏提取器中,加甲醇70 ml,置70℃水浴中回流提取8 h,提取液用甲醇定容至100rnL,提取和储存时注意避光。测定时,取此溶液5mL用甲醇稀释至25mL作为测试液。
3 实验条件
薄层层析条件:以聚酰胺薄膜为固定相,苯一甲醇一甲酸(10:5:1)为展开剂,上行一次展开,展距6cm。
荧光扫描条件:荧光反射法直线扫描,线性参数SX=3,光源为汞灯,二号滤光片,λEm=400 nm,λEx=306 rlm,狭缝0.4 mm×5 mm,△Y=0.04 mm,峰检出方式:峰面积法。
4 线性关系考察
准确吸取等体积0.lmg·mL- l反式白藜芦醇和0.1mg·m-l反式白藜芦醇苷对照品溶液混合,用甲醇稀释10倍后,取此溶液分别点样0.4,1.0,2.0,3.0,4.0 μL于同一聚酰胺薄膜上;同样,吸取等体积顺式白藜芦醇和顺式白藜芦醇苷对照品溶液混合,用甲醇稀释5倍后,分别点样0.4,1.0,2.0,3.0,4.0 L于另一聚酰胺薄膜上。按层析条件展开,按扫描条件扫描。以峰面积Y对相应的对照品的X(ng)进行线性回归,反式白藜芦醇及其糖苷和顺式白藜芦醇及其糖苷的回归方程分别为:
Y=207.86X+334.48 r=0.9972
Y=50.177X+88.085 r= 0.9991
Y=184.79X+164.94 r= 0.9996
Y=68.I48X+I68.27 r= 0.9982
线性范围分别为:2.00~20.0ng,2.00~20.0 ng,2.77~27.7 ng,2.32~23.2 ng。
5 稳定性考察
将有4种对照品斑点的聚酰胺薄膜分别在自然光和避光条件下存放,在l0h内每隔30 min扫描1次。结果表明,在避光条件下,薄膜上组分的扫描峰面积在l0h内稳定(RSD=1.2%~1.5%),不避光保存时,斑点的扫描峰面积下降。因此,操作中应注意避光进行。
6 精密度考察
6.1 同板精密度
取4种对照品的混合液,在同一聚酰胺薄膜上点同样的量6次,按测定条件进行展开和扫描,分别测定其峰面积,求得RSD为1.7%~2.3%。
6.2 异板精密度
取4种对照品的混合液,点同样的量于6块不同的聚酰胺薄膜上,按测定条件进行展开和扫描,分别测定其峰面积,求得RSD为2.1%~3.1%。
7 加样回收率试验
准确称取5份已知测定组分含量的虎杖粉,每份均为0.500 g,分别加人反式白藜芦醇对照品1.005 mg、反式白藜芦醇苷对照品10.002 mg,按样品供试液的制备 方法制备测试液,按层析条件展开,按扫描条件扫描,以对照品斑点定性,以外标法定量。测得反式白藜芦醇和反式白藜芦醇苷的平均回收率分别为97.1%(RSD=2.6%)和96.3%(RSD=2.5%)。
8 样品测定
准确吸取样品测试液1 μL与顺式及反式白黎芦醇及其糖苷的混合对照品,点样于同一聚酰胺薄膜上,按测定条件层析和扫描,以样品与混合对照品斑点的比移值及紫外灯下色斑特征定性,以外标法定量。对照品薄层扫描结果见图1,样品薄层扫描结果见图2,表1为样品测定结果。
-
+关注
私聊
-
yhl-87_
第29楼2009/11/06
薄层色谱法应用系列讲座-白藜芦醇及其糖苷异构体含量(23) 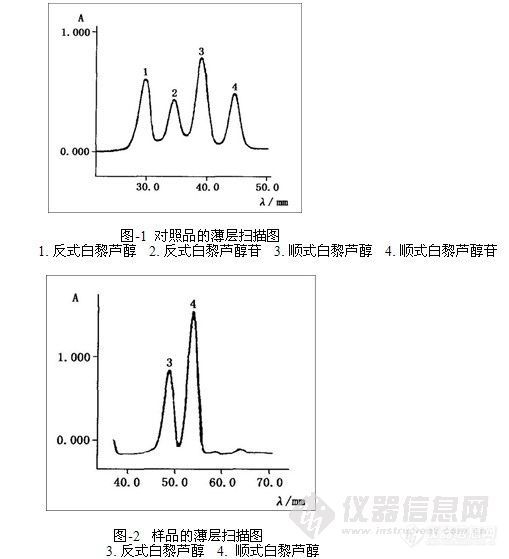
9 讨论
9.1 取对照品混合液点样于聚酰胺薄膜上,按层析条件展开后,用扫描仪进行各组分的斑点原位扫描,测得反式白藜芦醇和反式白藜芦醇苷的最大吸收波长分别为306 nm和318 nm,顺式白藜芦醇和顺式白藜芦醇苷则分别为290 nm和288 nm。实验中兼顾4种组分的吸收波长,选定的激光波长入Ex为306 nm,选择二号滤光片,发射光波长入Em为400 nm。
以硅胶GF254.作固定相,对多种展开剂试验,结果表明,硅胶G只能分离白藜芦醇与其糖苷,而不能分开它们的顺、反异构体。改以聚酰胺薄膜为固定相,同样对多种展开剂试验,发现采用苯一甲醇一甲酸(10:5:1)作展开剂,不仅分离度好,且各个斑点的比移值适中。故试验以聚酰胺薄膜作固定相,苯一甲醇一甲酸(l0:5:1)为展开剂层析分离。
9.2 本文首次采用聚酰胺薄膜作固定相,不仅能分离白藜芦醇与其糖苷,而且能区分它们的顺、反式异构体,因此本方法除可用于检测虎杖制品中的白藜芦醇含量外,还能检测虎杖制品在储存中发生的白藜芦醇及其糖苷的顺反异构体转化。实验采用荧光扫描测定,避免了使用紫外检测时其他组分的干扰。同时测定的灵敏度提高了近100倍。方法简便、快速、灵敏、测定成本低。
9.3 实验结果证明,在避光条件下提取和测定的4个虎杖样品中反式白藜芦醇和反式白藜芦醇带的含量分别为0.20%~0.22%和2.04%~2.14%,在此条件下虎杖提取液中没有可定量测定的顺式异构体。
-
+关注
私聊
-
yhl-87_
第30楼2009/11/06
薄层色谱法应用系列讲座(24)-丹磺酰化氨基酸聚合物合成性能研究
印记丹磺酰化氨基酸聚合物固定相的合成及色谱性能研究
摘 要 目的:建立分子印记的薄层板拆分对映体的方法。
方法:以丹磺酰化-L -苯丙氨酸(dansyl –L- phenylalanine,Dns-L-phe)为模板分子,分别以甲基丙烯酸(methacrylic acid,MAA)为单功能单体、MAA与2-乙烯基吡啶(2- vinylpyridine,2-Vpy)为双功能单体,采用本体聚合方法制备了2种模板聚合物。将聚合物与自制煅石膏混合制成分子印记的薄层板,结合展开剂的优化,对丹磺酰化-DL-苯丙氨酸(Dns-DL-phe)对映体进行拆分研究。
结果:当展开剂为乙腈-乙酸(9.9:0.1)时,Dns-DL- phe在2种印记薄层板上都能得到完全分离,分离因子分别为1.7和2.2。
结论:基于分子印记技术的薄层色谱提供了一种潜在、快速、有效地拆分对映体的方法。
本文由徐莉1,何建峰2,刘岚2,邓芹英2(1,华南农业大学化学系;2.中山大学化学系)等研究人员共同完成的国家自然科学基金资助项目的部分内容,他们用自制的2种模板聚合物,能快速有效地拆分对映体。现全文介绍如下
前 言
分子印记技术是一门正在发展中的新兴技术。有关分子印记聚合物(molecularly imprinted polymer,MIP)固定相用于高效液相色谱分离对映体的研究较多,而将MIP用于薄层分离对映体的研究[1-3] 较少,国内还未见文献报道。薄层色谱具有简单、直接、快速等诸多优点,本文以丹磺酰化-L -苯丙氨酸(Dns-L - phe)为模板分子,分别以甲基丙烯酸(MAA)为单功能单体、MAA与2-乙烯基吡啶(2 - Vpy)为双功能单体,以乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate,EDMA)为交联剂,采用本体聚合方法制备了2种MIP。将MIP与自制的煅石膏混合制成分子印记的薄层板,结合展开剂的优化,使丹磺酰化-DL -苯丙氨酸(Dns-DL-phe)对映体得到完全分离。
实 验
1 实验部分
1.1 原料、试剂与仪器
Dns-L-phe,丹磺酰氯(Dns- Cl),Sigma公司产品。DL -苯丙氨酸(DL - phe),第二军医大学朝晖制药厂。甲基丙烯酸(MAA,上海化学试剂公司),二乙烯基吡啶(Acros公司产品),乙二醇二甲基丙烯酸酯(EDMA,江苏安利化工厂,用1 mol·L-1 饱和氢氧化钠溶液洗涤,再用饱和氯化钠溶液洗涤,最后用无水硫酸镁干燥),以上试剂需减压蒸馏除去阻聚剂。硫酸钙由本实验室自制。其他试剂为分析纯。
德国Brucker公司的VECTOR型傅立叶变换红外光谱仪,日本电子ZSM - 6300F型场发射电子扫描显微镜,日本岛津TGA-50型热重分析仪,C-86型恒温水浴锅(广州越秀医疗器械厂)。
1.2 丹磺酰化-DL一苯丙氨酸的制备
参照文献[4],将适量的DL- phe溶于0.1 mmol·L-1 碳酸氢钠溶液中,取此溶液1 mL与15 mmol·L-1 丹磺酰氯的无水溶液避光反应2 h。反应液用乙醚萃取,除去过量的丹磺酰氯。水层用0.2 mmol·L-1 盐酸调pH为4,用乙酸乙酯萃取,乙酸乙酯层用无水硫酸镁干燥后,蒸去乙酸乙酯,残余物用甲醇配成浓度为1 mg·mL-1 的溶液,供TLC拆分用。
1.3 印记丹磺酰化-L-苯丙氨酸的分子烙印聚合物的制备
称取模板分子Dns- L - phe 0.379 g(1 mmol),MAA 0.344 g(4 mmoL)或MAA
0.172 g和2 - Vpy 0.21 g(各为2 mmoL)于10 mL乙腈中,放置于10 mL锥形瓶中,超声并通氮气5 min,然后加人交联剂EDMA 3.964 g(20 mmoL)、偶氮二异丁腈30 mg,转人50 mL安瓿瓶中,超声通氮气5 min,真空脱气5 min后封管,置50℃恒温水浴锅中反应24 h,得到块状混合物。将得到的块状混合物研碎,过200目筛,先在常温下用乙腈搅拌24 h,然后分别用9:1的甲醇与乙酸和9:1的乙腈与乙酸抽提数天,再用甲醇洗涤。用丙酮反复沉降,除去细小粉末后在50℃真空干燥24 h。单功能单体聚合物和双功能单体聚合物分别用MIP1、MIP2表示;非模板聚合物的制备除不加模板分子,其余方法同上。对应的空白聚合物分别用NIP1、NIP2表示。
1.4 薄层板的制备
称取自制煅石膏和模板聚合物各0.8 g,于研钵中研磨均匀,加水6 mL混合,迅速均匀涂布于已备好的干净载玻片上(2.5 cm×7.5 cm),制成厚度均匀、表面平滑的薄层,室温下水平晾干,取出后置于干燥器中备用,薄层板的厚度约0.25 mm。同时制备一定量的非模板聚合物薄层板,以便进行对照分析。
1.5 色谱分离过程
将Dns- L- phe、Dns- DL-phe溶于甲醇,配成浓度约为1 mg·mL-l 的溶液。用毛细管在距板边缘1 cm处点样,室温下在饱和的层析缸中上行展开。展开剂分别为含有不同乙酸浓度(0%,1%,5%)的氯仿溶液、乙腈溶液、甲醇溶液。在紫外灯(λ=254 nm或λ=365 nm)下观察样品斑点的分离情况。
2 结果与讨论
2.1 聚合物的结构表征
2.1.1 红外光谱(IR)
将所得的上述聚合物进行红外光谱表征(溴化钾压片法)。IR谱图表明,所有聚合物有明显的羟基、羰基、酯基的特征吸收峰,以MIP1为例,3449 cm-1 附近强而宽的峰归属于一CO0H伸缩振动,1729 cm -1 归属于C=O的伸缩振动,1456,1391 cm -1 分别为-CH2和-CH3弯曲振动峰。MIP2和NIP2的谱图中,除了上述特征吸收峰外,在1639,1637 cm -1 的处吸收峰为吡啶的VC=N吸收,这是由于与]υC= C的吸收峰重叠而使峰强度明显增强。
2.1.2 热重分析(TG)
将所得的模板聚合物用热重分析仪进行测定。升温速度:10℃·min -1;记录温度:室温至730℃;气氛:空气。结果表明,MIP在240℃以下对热稳定,500℃左右基本分解完全。
2.1.3 电镜扫描(SEM)
对印记模板的MIP和空白聚合物进行电镜扫描并观察其表面形貌,可以看出,印记有模板分子的聚合物表面有许多微孔,而空白聚合物表面平滑,表明聚合物对模板分子产生了明显的印迹。扫描电镜见图1。

2.2 分子印记聚合物的制备
本实验分别采用单功能单体(MAA)和双功能单体(MAA+2 -Vpy)来印记模板分子。由于Dns -L - phe在紫外灯下有荧光,而模板分子的存在不仅会降低MIP的识别能力,而且还会干扰紫外检测,因此需把模板分子尽可能洗去。实验证明,尽管模板分子不可能完全去除,但微量的模板分子存在不影响紫外检测。
2.3 薄层板的制备
由于单纯附着在玻璃板上的MIP容易开裂,因此选择合适的粘合剂是关键。实验中采用常用的粘合剂羧甲基纤维素钠制成的板,机械性能差,容易开裂。而加人自制粘合剂煅石膏与MIP等量混合,制得的薄层板表面光滑平整,粘合性能与机械性能大大提高。此外,煅石膏的加入也降低了薄层板的紫外吸收背景,有利于样品斑点的检测。
2.4 薄层色谱的拆分研究
将Dns -DL - phe和Dns - L - phe的甲醇溶液,用毛细管点样,分别用上述3种含有不同乙酸含量(0%,1%,5%)的氯仿、乙腈、甲醇为展开剂,在展开剂饱和的层析缸中室温上行展开,同时用空白板进行对照。拆分结果见表1。
由表可知,当展开剂为乙腈-乙酸(9.9:0.1)时,Dns- DL- phe在2种MIP板上都能得到拆分,见图2。其中MIP2聚合物固定相的拆分能力略大于MIPI聚合物固定相,分离因子分别为2.2和1.7。可能的原因是双功能单体能更好地和模板分子产生相互作用,吡啶基上的N原子不仅是氢键接受体,而且吡啶环与本丙氨酸的苯环也有π-π作用,从而增加了识别作用位点。2种非模板聚合物对Dns -DL - phe均没有表现出拆分能力,与印记聚合物薄层板相比,溶质在非模板聚合物薄层板上的保留明显降低,以MIP2和NIP2对溶质的拆分为例,当展开剂体系为乙腈一乙酸(9.9:0.1)时,溶质在NIP2薄层板上的Rf (L) 和Rf (D) 均为0.56,而在MIP2薄层板上保留很强,其Rf (L)和Rf (D)分别为0.09,0.20。
对展开剂的摸索实验发现,乙腈的分离效果明显好于氯仿和甲醇,在展开剂中加人少量乙酸,会提高样品斑点的相对比移值,并使斑点圆整、清晰。但乙酸加人量不宜太大,否则会使TLC板不稳定,容易脱落。对MIP2板来说,因聚合物中含有碱性的吡啶基,也不宜使用酸性太强的洗脱剂。
