-
+关注
私聊
-

yhl-87_
第31楼2009/11/06
薄层色谱法应用系列讲座(25)-分子印记的薄层板拆分对映体方法
印记丹磺酰化氨基酸聚合物固定相的合成及色谱性能研究
摘 要 目的:建立分子印记的薄层板拆分对映体的方法。
方法:以丹磺酰化-L -苯丙氨酸(dansyl –L- phenylalanine,Dns-L-phe)为模板分子,分别以甲基丙烯酸(methacrylic acid,MAA)为单功能单体、MAA与2-乙烯基吡啶(2- vinylpyridine,2-Vpy)为双功能单体,采用本体聚合方法制备了2种模板聚合物。将聚合物与自制煅石膏混合制成分子印记的薄层板,结合展开剂的优化,对丹磺酰化-DL-苯丙氨酸(Dns-DL-phe)对映体进行拆分研究。
结果:当展开剂为乙腈-乙酸(9.9:0.1)时,Dns-DL- phe在2种印记薄层板上都能得到完全分离,分离因子分别为1.7和2.2。
结论:基于分子印记技术的薄层色谱提供了一种潜在、快速、有效地拆分对映体的方法。
本文由徐莉1,何建峰2,刘岚2,邓芹英2(1,华南农业大学化学系;2.中山大学化学系)等研究人员共同完成的国家自然科学基金资助项目的部分内容,他们用自制的2种模板聚合物,能快速有效地拆分对映体。现全文介绍如下
前 言
分子印记技术是一门正在发展中的新兴技术。有关分子印记聚合物(molecularly imprinted polymer,MIP)固定相用于高效液相色谱分离对映体的研究较多,而将MIP用于薄层分离对映体的研究[1-3] 较少,国内还未见文献报道。薄层色谱具有简单、直接、快速等诸多优点,本文以丹磺酰化-L -苯丙氨酸(Dns-L - phe)为模板分子,分别以甲基丙烯酸(MAA)为单功能单体、MAA与2-乙烯基吡啶(2 - Vpy)为双功能单体,以乙二醇二甲基丙烯酸酯(ethylene glycol dimethacrylate,EDMA)为交联剂,采用本体聚合方法制备了2种MIP。将MIP与自制的煅石膏混合制成分子印记的薄层板,结合展开剂的优化,使丹磺酰化-DL -苯丙氨酸(Dns-DL-phe)对映体得到完全分离。
实 验
1 实验部分
1.1 原料、试剂与仪器
Dns-L-phe,丹磺酰氯(Dns- Cl),Sigma公司产品。DL -苯丙氨酸(DL - phe),第二军医大学朝晖制药厂。甲基丙烯酸(MAA,上海化学试剂公司),二乙烯基吡啶(Acros公司产品),乙二醇二甲基丙烯酸酯(EDMA,江苏安利化工厂,用1 mol·L-1 饱和氢氧化钠溶液洗涤,再用饱和氯化钠溶液洗涤,最后用无水硫酸镁干燥),以上试剂需减压蒸馏除去阻聚剂。硫酸钙由本实验室自制。其他试剂为分析纯。
德国Brucker公司的VECTOR型傅立叶变换红外光谱仪,日本电子ZSM - 6300F型场发射电子扫描显微镜,日本岛津TGA-50型热重分析仪,C-86型恒温水浴锅(广州越秀医疗器械厂)。
1.2 丹磺酰化-DL一苯丙氨酸的制备
参照文献[4],将适量的DL- phe溶于0.1 mmol·L-1 碳酸氢钠溶液中,取此溶液1 mL与15 mmol·L-1 丹磺酰氯的无水溶液避光反应2 h。反应液用乙醚萃取,除去过量的丹磺酰氯。水层用0.2 mmol·L-1 盐酸调pH为4,用乙酸乙酯萃取,乙酸乙酯层用无水硫酸镁干燥后,蒸去乙酸乙酯,残余物用甲醇配成浓度为1 mg·mL-1 的溶液,供TLC拆分用。
1.3 印记丹磺酰化-L-苯丙氨酸的分子烙印聚合物的制备
称取模板分子Dns- L - phe 0.379 g(1 mmol),MAA 0.344 g(4 mmoL)或MAA
0.172 g和2 - Vpy 0.21 g(各为2 mmoL)于10 mL乙腈中,放置于10 mL锥形瓶中,超声并通氮气5 min,然后加人交联剂EDMA 3.964 g(20 mmoL)、偶氮二异丁腈30 mg,转人50 mL安瓿瓶中,超声通氮气5 min,真空脱气5 min后封管,置50℃恒温水浴锅中反应24 h,得到块状混合物。将得到的块状混合物研碎,过200目筛,先在常温下用乙腈搅拌24 h,然后分别用9:1的甲醇与乙酸和9:1的乙腈与乙酸抽提数天,再用甲醇洗涤。用丙酮反复沉降,除去细小粉末后在50℃真空干燥24 h。单功能单体聚合物和双功能单体聚合物分别用MIP1、MIP2表示;非模板聚合物的制备除不加模板分子,其余方法同上。对应的空白聚合物分别用NIP1、NIP2表示。
1.4 薄层板的制备
称取自制煅石膏和模板聚合物各0.8 g,于研钵中研磨均匀,加水6 mL混合,迅速均匀涂布于已备好的干净载玻片上(2.5 cm×7.5 cm),制成厚度均匀、表面平滑的薄层,室温下水平晾干,取出后置于干燥器中备用,薄层板的厚度约0.25 mm。同时制备一定量的非模板聚合物薄层板,以便进行对照分析。
1.5 色谱分离过程
将Dns- L- phe、Dns- DL-phe溶于甲醇,配成浓度约为1 mg·mL-l 的溶液。用毛细管在距板边缘1 cm处点样,室温下在饱和的层析缸中上行展开。展开剂分别为含有不同乙酸浓度(0%,1%,5%)的氯仿溶液、乙腈溶液、甲醇溶液。在紫外灯(λ=254 nm或λ=365 nm)下观察样品斑点的分离情况。
2 结果与讨论
2.1 聚合物的结构表征
2.1.1 红外光谱(IR)
将所得的上述聚合物进行红外光谱表征(溴化钾压片法)。IR谱图表明,所有聚合物有明显的羟基、羰基、酯基的特征吸收峰,以MIP1为例,3449 cm-1 附近强而宽的峰归属于一CO0H伸缩振动,1729 cm -1 归属于C=O的伸缩振动,1456,1391 cm -1 分别为-CH2和-CH3弯曲振动峰。MIP2和NIP2的谱图中,除了上述特征吸收峰外,在1639,1637 cm -1 的处吸收峰为吡啶的VC=N吸收,这是由于与]υC= C的吸收峰重叠而使峰强度明显增强。
2.1.2 热重分析(TG)
将所得的模板聚合物用热重分析仪进行测定。升温速度:10℃·min -1;记录温度:室温至730℃;气氛:空气。结果表明,MIP在240℃以下对热稳定,500℃左右基本分解完全。
2.1.3 电镜扫描(SEM)
对印记模板的MIP和空白聚合物进行电镜扫描并观察其表面形貌,可以看出,印记有模板分子的聚合物表面有许多微孔,而空白聚合物表面平滑,表明聚合物对模板分子产生了明显的印迹。扫描电镜见图1。

-
+关注
私聊
-
yhl-87_
第32楼2009/11/06
薄层色谱法应用系列讲座(25)(下)-分子印记的薄层板拆分对映体方法
2.2 分子印记聚合物的制备
本实验分别采用单功能单体(MAA)和双功能单体(MAA+2 -Vpy)来印记模板分子。由于Dns -L - phe在紫外灯下有荧光,而模板分子的存在不仅会降低MIP的识别能力,而且还会干扰紫外检测,因此需把模板分子尽可能洗去。实验证明,尽管模板分子不可能完全去除,但微量的模板分子存在不影响紫外检测。
2.3 薄层板的制备
由于单纯附着在玻璃板上的MIP容易开裂,因此选择合适的粘合剂是关键。实验中采用常用的粘合剂羧甲基纤维素钠制成的板,机械性能差,容易开裂。而加人自制粘合剂煅石膏与MIP等量混合,制得的薄层板表面光滑平整,粘合性能与机械性能大大提高。此外,煅石膏的加入也降低了薄层板的紫外吸收背景,有利于样品斑点的检测。
2.4 薄层色谱的拆分研究
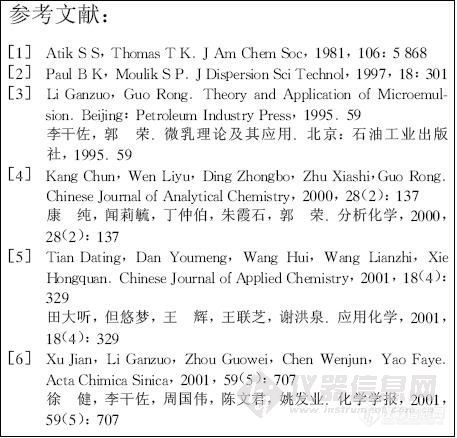
将Dns -DL - phe和Dns - L - phe的甲醇溶液,用毛细管点样,分别用上述3种含有不同乙酸含量(0%,1%,5%)的氯仿、乙腈、甲醇为展开剂,在展开剂饱和的层析缸中室温上行展开,同时用空白板进行对照。拆分结果见表1。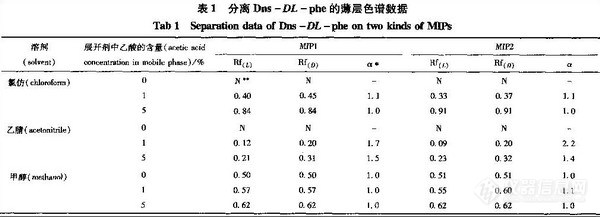
由表可知,当展开剂为乙腈-乙酸(9.9:0.1)时,Dns- DL- phe在2种MIP板上都能得到拆分,见图2。其中MIP2聚合物固定相的拆分能力略大于MIPI聚合物固定相,分离因子分别为2.2和1.7。可能的原因是双功能单体能更好地和模板分子产生相互作用,吡啶基上的N原子不仅是氢键接受体,而且吡啶环与本丙氨酸的苯环也有π-π作用,从而增加了识别作用位点。2种非模板聚合物对Dns -DL - phe均没有表现出拆分能力,与印记聚合物薄层板相比,溶质在非模板聚合物薄层板上的保留明显降低,以MIP2和NIP2对溶质的拆分为例,当展开剂体系为乙腈一乙酸(9.9:0.1)时,溶质在NIP2薄层板上的Rf (L) 和Rf (D) 均为0.56,而在MIP2薄层板上保留很强,其Rf (L)和Rf (D)分别为0.09,0.20。
对展开剂的摸索实验发现,乙腈的分离效果明显好于氯仿和甲醇,在展开剂中加人少量乙酸,会提高样品斑点的相对比移值,并使斑点圆整、清晰。但乙酸加人量不宜太大,否则会使TLC板不稳定,容易脱落。对MIP2板来说,因聚合物中含有碱性的吡啶基,也不宜使用酸性太强的洗脱剂。

-
+关注
私聊
-
yhl-87_
第33楼2009/11/06
薄层色谱法应用系列讲座(26)-TLC/HPLC复方氨酚烷胺片质量标准研究
下列资料由“仪器信息网”负责搜集整理,版权所有。请勿复制粘贴,否则视为侵权
复方氨酚烷胺片TLC/HPLC质量标准的研究
摘 要 目的:对复方氨酚烷胺片进行质量标准的研究。
方法:采用薄层色谱法对人工牛黄中贝斯素进行定性鉴别,以异辛烷一甲苯-正丁醇一冰乙酸一乙醇一水(1:4:4:2:1:1)为展开剂。采用高效液相色谱法对人工牛黄中胆红素进行含量测定,采用Kromasil C18色谱柱(4.6 mm×250 mm,5 μm),流动相为甲醇一氯仿-l%磷酸溶液(70:24:6),检测波长为450 nm,流速为1.0 mL·min -l。
结果:薄层色谱鉴别方法专属性强。含量测定胆红素在2.45~17.2 μg·mL -l 范围呈良好的线性关系,r=0.9996,平均回收率为100.6%,RSD为1.5%(n =9),重复性试验RSD为1.5%(n=9),
结论:所建立的方法可准确、快速地进行定性、定量检测,可用于该制剂的质量控制。
本文由湖南省邵阳市药品检验所的梁仪容,胡剑,吴活龙,乐立源,王伟姣等研究人员共同完成,提供了许多实践经验值得参考。现全文介绍如下。
前 言
复方氨酸烷胺片是由对乙酰氨基酚、盐酸金刚烷胺、人工牛黄、咖啡因和马来酸氯苯那敏组成的复方制剂[1],具有解热、镇痛、抗炎、抗过敏等作用,是临床广泛使用的抗感冒药,该制剂收载于《国家药品标准》化学药品地升国标第十六册中,但这一质量标准无该处方组成中人工牛黄主要成分的定性定量分析。我们采用高效液相色谱法测定了人工牛黄中所含有效成分胆红素的含量[2~3],用薄层色谱法对人工牛黄中贝斯素进行鉴别。结果表明该方法简便,结果准确,专属性强,对控制本品的质量具有重要意义。
实 验
1 仪器与试药
美国SP高效液相色谱仪。日本岛津UV -2201紫外分光光度计。
对照品:胆红素(供含量测定用,批号10077 -0301)、贝斯素(供鉴别用批号0267 -9701),均由中国药品生物制品检定所提供;缺贝斯素的人工牛黄由厂方提供,薄层色谱用硅胶G(青岛海洋化工有限公司);甲醇、氯仿均为色谱纯,水为重蒸馏水,其他试剂均为分析纯。复方氨酚烷胺片由6个厂家(A,B,C,D,E,F)提供的10批样品;阴性样品:人工牛黄用缺贝斯素的人工牛黄代替,各药物按处方量依法配制。阳性样品:按处方量依法配制。
2 方法与结果
2.1 贝斯素的薄层色谱鉴别
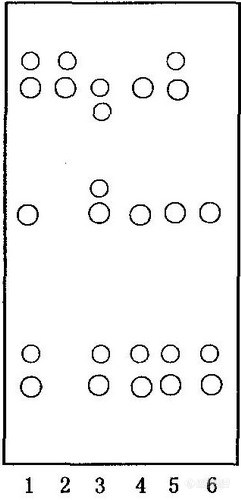
取本品2片,研细,加水20mL,超声处理10 min,滤过,滤液加人预先处理好的聚酰胺柱(14~30目,内径1.5 cm,装柱高10 cm)上,用水80 mL洗脱,弃去水液,再以甲醇40 mL洗脱,收集甲醇洗脱液,水浴蒸干,残渣加甲醇2 mL使溶解,作为供试品溶液;分别取阴性样品和阳性样品各相当于约2片量,同法操作,制得阴性对照液和阳性对照液;另取贝斯素对照品,加甲醇制成每1 mL含2 mg的溶液,作为对照品溶液。照薄层色谱法[4],分别吸取上述4种溶液各2~4μL,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以异辛烷一甲苯一正丁醇-冰乙酸一乙醇一水(1:4:4:2:1:1)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105℃加热约5 min,置紫外光灯365 nm下检视,供试品色谱中,在与对照品色谱相应的位置上,显3个或3个以上相同颜色的荧光主斑点。阴性对照无干扰。结果见图1。
阳性样品 2. 阴性对照 3. 贝斯素对照品 4.、5、6. 不同的被测样品
2.2 胆红素含量测定
2.2.1 色谱条件
色谱柱Kromasil C18(4.6 mm×250 mm,5 μm);流动相:甲醇-氯仿-1%磷酸溶液(70:24:6),检测波长:450 nm;流速1.0 mL·min -1,进样量10μL。
按上述色谱条件试验,胆红素色谱峰可达基线分离,且分离效果良好,R>1.5;按胆红素峰计算,理论板数在4000以上。取缺人工牛黄的阴性样品液进行测定,在与胆红素对照品峰相应位置上无峰,说明阴性无干扰,所得结果见图-2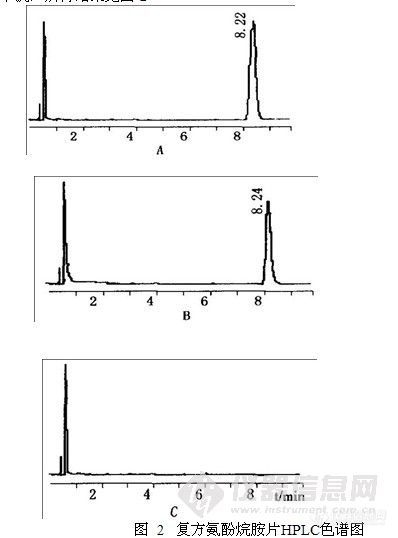
A 胆红素对照品 B 供试品 C 阴性对照品
2.2.2 对照品溶液的制备
精密称取胆红素对照品5 mg,置50 mL棕色量瓶中,加溶剂氯仿-甲醇一磷酸盐缓冲液pH 7.8~8.0(70:10:0.4)适量,超声使溶解并稀释至刻度,摇匀,精密量取2 mL,置50 mL棕色量瓶中加上述溶剂至刻度即得。
2.2.3 供试品溶液的制备
取本品20片,经研细,精密称取适量(约相当于人T牛黄30 mg),置50 mL棕色量瓶中,加入溶剂氯仿一甲醇一磷酸盐缓冲液pH 7.8~8.0(70:l0:0.4)40 mL,超声提取20min,放冷,加上述溶剂至刻度,摇匀,滤过,取续滤液作为供试品溶液。
2.2.4 线性关系考察
精密称取胆红素对照品约5 mg置100 mL棕色量瓶中,加溶剂氯仿-甲醇一磷酸盐缓冲液pH 7.8~8.0(70:10:0.4)适量使溶解并稀释至刻度,分别取以上配制的对照品溶液0.5,1.0,2.0,2.5,3.0,3.5 mL置10 mL棕色量瓶中,分别加上述溶剂至刻度,摇匀,在上述色谱条件下进行测定,分别进样10μL,记录色谱峰面积,以胆红素浓度(C)为横座标,峰面积(A)为纵座标,经统计得回归方程为:
A= 136827C -52952 r =0.9996
结果表明胆红素对照品在2.45~17.2μg·mL -1的浓度范围内有良好的线性关系。


-
+关注
私聊
-
yhl-87_
第34楼2009/11/06
薄层色谱法应用系列讲座(26)(下)-TLC/HPLC复方氨酚烷胺片质量标准研究
2.2.5 稳定性试验
分别取按上述方法配制的同一份对照品和供试品溶液,分别在0,1,2,3,4,5,7,8,16和0,1,2,3,4,5,6 h进样10 μL,测定峰面积积分值,结果:峰面积积分值的RSD分别为0.95%和0.85%,结果表明对照品溶液和供试品溶液分别在16 h和6 h内稳定。
2.2.6 精密度考察 重复性试验:
取同一批供试品,依法配制成80%,100%,120%3种浓度的供试品溶液9份,按上述色谱条件,每次进样10μL,按外标法计算,结果胆红素的含量分别为56.77,57.87,57.78,56.87,58.74,56.92,58.55,59.03,58.74 μg·片 -l, RSD=1.5%。
重现性试验:取同一批供试品,经湖南省株洲市药品检验所进行重现性试验,结果胆红素的含量分别为 62.26,59.35,59.04,60.34,59.34,59.28,61.76,60.00,60.43μg·片-l,RSD=1.9%。从重复性试验和重现性试验结果表明,该方法具有良好的精密度。
2.2.7 回收率考察 回收率试验:
分别称取约一片重的阴性对照品粉末9份,分别精密加人胆红素170μg,255μg,542μg 3种浓度级,每浓度级配制3份,照供试品溶液制各项下的方法操作,按上述色谱条件,测定胆红素的含量,计算回收率,结果平均回收率为100.6%,RSD为1.5%。
加样回收率试验:取已测定准确含量的3批供试品,每批供试品称取3个浓度级,并分别精密加人一定量的胆红素对照品,照供试品溶按各项下的方法操作,按上述色谱条件,测定胆红素的含量,计算回收率,结果平均回收率为100.2%,RSD为1.9%。
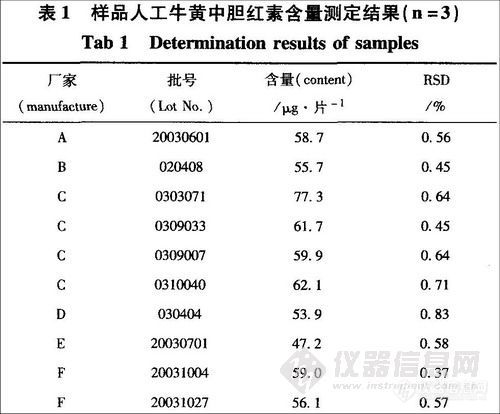
2.2.8 样品人工牛黄中胆红素含量测定
按对照品溶液和供试品溶液制各项下的方法配制对照品溶液和l0批供试品溶液,按上述色谱条件,每次进样l0μL,记录色谱图,按外标法计算,结果见表1。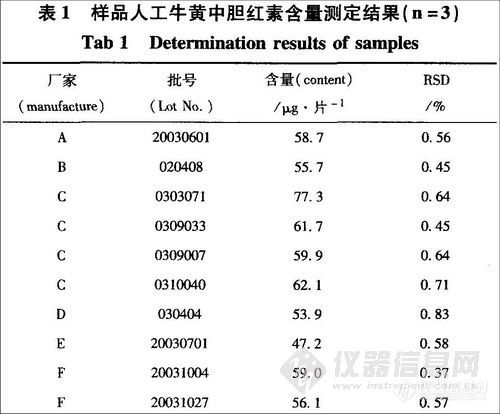
3 讨 论
复方氨酚烷胺片原质量标准中,仅对人工牛黄中胆酸进行了化学鉴别,我们增加人工牛黄中主要成分贝斯素的薄层色谱鉴别,对检出不含人工牛黄的此类假药,有重要意义。
经查阅有关资料和文献[5],胆红素是一个大分子量(584.65)化合物,溶于苯、氯仿等。干燥时稳定,但在酸、碱溶液中易被氧化,见光加速,故在含量测定过程中宜避光操作,采用棕色量瓶定容。
测定波长的选择:取胆红素对照品用氯仿稀释成每mL含50 μg的溶液,照分光光度法,在300 nm~500nm波长范围扫描,结果在452nm波长处有最大吸收,与文献报道相符,所以测定波长定于450 nm处。
选择合适的流动相:在相当一部分资料中[6],可见到用HPLC法分离胆红素的流动相多以甲醇、乙腈和1%醋酸配比组成,经试验胆红素对照品能分出3个峰(为同分异构体),但我们至今还无法找到标示主峰含量的胆红素对照品。同时供试品溶液在胆红素峰前面有一个大的溶剂拖尾峰,使胆红素不能基线分离,于是考虑加人氯仿。经反复试验,把流动相调整为甲醇一氯仿-1%磷酸溶液(70:24:6),结果获得满意效果,峰形好,胆红素峰可达基线分离,R>1.5,理论板数在4000以上,tR=8.2 min。
提取方法的选择:我们把提取溶剂的种类、提取溶剂的体积和提取时间作为考察因素,每个因素选3个水平进行正交试验,结果以氯仿一甲醇(5:1)为提取溶剂,加人40 mL,超声20min为最佳提取方案。但胆红素对照品在该溶剂体系中不稳定,经测定2 h下降5%。而胆红素在供试品溶液中较稳定,由此得到启迪,我们在溶剂中加人微量的缓冲液,把溶解胆红素对照品的溶剂改为氯仿一甲醇一磷酸盐缓冲液pH7·8~8.0(70:10:0.4),获得满意效果。对照品胆红素峰面积值在16 h内稳定,供试品胆红素峰面积值在6 h内稳定。
致谢:分析方法的验证由湖南省株州市药检所协作完成,在此致谢。


-
+关注
私聊
-
yhl-87_
第35楼2009/11/06
薄层色谱法应用系列讲座(27)-对乳安颗粒的质量控制
薄层色谱对乳安颗粒质量控制的研究
摘 要
目的:制定乳安颗粒质量控制方法。
方法:采用薄层色谱法对处方中的柴胡、当归、浙贝母、连翘、向术、茯苓、延胡索进行定性鉴别;采用高效液相色谱法对处方中自芍中的芍药苷进行含量测定。
结果:芍药苷在0.16~1.04μg范围内呈良好的线性关系(r = 0.9998).加样回收率为98.9%~101.6%。
结论:本方法能对乳安颗粒进行定性、定量检测,结果准确可靠。
本文由山西大学化学化工学院药学系的王光丽,王爱娜,秦雪梅,张丽增,郭小青等老师们共同完成,全文介绍如下。
前 言
乳安颗粒是由柴胡、当归、白芍、延胡索等16味药经提取制成的颗粒剂。具有壮腰健肾、养血滋阴的作用。该药成分复杂,为了有效地控制本品的内在质量,对处方中的柴胡、当归、浙贝母、连翘、白术、茯苓、延胡索进行薄层定性鉴别,并改进试验条件,对其中具有抗炎、抗溃疡、镇痛等作用[l]的有效成分芍药苷采用高效液相色谱法进行了含量测定,方法简便、灵敏、可靠。
实 验
1 仪器与试药
Waters高效液相色谱仪,Breeze色谱工作站,1525泵;2487紫外检测器。乙腈为色谱纯;水为重蒸水;其它试剂均为分析纯。芍药苷(供含测用)、贝母素甲、贝母素乙、连翘苷、延胡索乙素(均为供鉴别用)五种对照品以及柴胡、当归、白术、茯苓四种对照药材均购自中国药品生物制品检定所;乳安颗粒为山西傅山药业提供。
2 薄层定性鉴别
2.1 柴胡的TLC鉴别
取本品6 g,加甲醇100 mL,加热回流1 h,滤过,滤液蒸干,残渣加20mL沸水使溶解,加到D101大孔树脂柱(内径1.5cm长20 cm)上,用水洗脱至洗脱液近无色,再分别以20%,40%,60%,80%乙醇各100 mL洗脱,收集80%乙醇洗脱液,蒸干,用甲醇1 ml使溶解,作为供试品溶液。另取柴胡对照药材0.5 g加甲醇50 mL,同法制成对照药材溶液1 mL。再取缺柴胡的阴性样品5 g,同法制得阴性对照溶液。分别吸取上述3种溶液各10 μL,分别点于同一硅胶G薄层板上,以醋酸乙酯一乙醇一水(8:2:1)展开,取出晾干,喷以2%对二甲氨基苯甲醛的40%硫酸溶液,60℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱相应的位置上,显相同的红色斑点,阴性对照无相应斑点(图1 -A)。
2.2 当归的TLC鉴别[2、3]
取本品5 g,加硅藻土0.5 g,研细,加乙醇20mL,浸渍1 h,超声处理20min,滤过,滤液浓缩成1 mL作为供试品溶液。另取当归对照药材0.2 g,加乙醇l0 mL同法制成对照药材溶液。再取缺当归阴性样品5g ,同法制得阴性对照溶液。分别吸取供试品溶液、阴性对照溶液各10μL,对照药材溶液2μL,点于同一硅胶G薄层板上,以正己烷一醋酸乙酯(9:1)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同的蓝色荧光斑点,阴性对照无相应斑点(图1 -B)。
2.3 浙贝母的TLC鉴别
取本品10 g,加硅藻±1g,研细,加50%乙醇60 mL,加热回流1 h,放冷,滤过,滤液蒸干,残渣以0.5%盐酸30 mL溶解,离心,上清液以浓氨试液调pH至9~10,用氯仿萃取2次,每次40 mL,萃取液挥干,残渣以氯仿1 mL使溶解,作为供试品溶液。另取贝母素甲和贝母素乙对照品,加乙醇制成1 mL各含2 mg的对照品溶液,再取缺浙贝母阴性样品10 g,同法制得阴性对照溶液。分别吸取上述4种溶液各10 μL,点于同一硅胶G薄层板上,以醋酸乙酯一甲醇-浓氨试液(17:2:1)为展开剂,展开,取出,晾干,喷以改良碘化铋钾试液显色。供试品色谱中,在与对照品色谱相应的位置上,显相同的橙红色斑点,阴性对照无相应斑点(图1 -C)。
2.4 连翘的TLC鉴别
收集“2.1”项下40%乙醇洗脱液,蒸干,以甲醇2 mL使溶解,作为供试品溶液。另取连翘苷对照品,加甲醇制成I mL含2 mg的对照品溶液。再取缺连翘阴性样品6 g,同法制得阴性对照溶液。分别吸取上述3种溶液各10 μL,点于同一硅胶G薄层板上,以氯仿一甲醇(5:1)为展开剂,展开,取出,晾干,喷以硫酸一乙醇(l:4),晾干,105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同的浅蓝色斑点,阴性对照无相应斑点(图1 -D)。
2.5 白术的TLC鉴别
取本品3 g,加硅藻±0.5g,研细,加正己烷15 mL,浸渍2 h,超声处理20min,滤过,滤液浓缩成1 mL作为供试品溶液。另取白术对照药材0.5 g,加正己烷5 ml,同法制成对照药材溶液。再取缺白术阴性样品3 g,同法制得阴性对照溶液。分别吸取供试品溶液、阴性对照溶液各l0μL,,对照药材溶液5 μL,点于同一硅胶G薄层板上,以石油醚(60~90℃)一醋酸乙酯(50:1)为展开剂,展开,取出,晾干,喷以5%香草醛硫酸溶液,供试品色谱中,在与对照药材色谱相应的位置上,显相同的浅蓝色斑点,阴性对照无相应斑点(图1 -E)。
2.6 茯苓的TLC鉴别
取本品12 g,加硅藻±2g,研细,加乙醚100 mL,加热回流2 h,滤过,滤液挥干,残渣以无水乙醇1 mL使溶解,作为供试品溶液。另取茯苓对照药材0.5 g,加乙醚50 mL同法制成对照药材溶液。再取缺茯苓阴性样品12 g,同法制得阴性对照溶液,分别吸取上述3种溶液各10 μL,点于同一硅胶GF254薄层板上,以苯一醋酸乙酯(16:7)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。供试品色谱中,在与对照药材色谱相应的位置上,显相同的浅蓝色荧光斑点,阴性对照无相应斑点(图I -F)。
2.7 延胡索的TLC鉴别
取本品3 g,加硅燥土0.5 g,研细,加乙醇50 mL,加热回流1 h,放冷,滤过,滤液蒸干,残渣加水50 mL,分次转移至分液漏斗中,加浓氨试液使呈碱性,用乙醚提取3次,每次20mL,合并乙醚提取液,挥去乙醚,残渣加乙醇1mL使溶解,作为供试品溶液。另取延胡索乙素对照品,加乙醇制成每1 mL含1 mg的溶液,作为对照品溶液。再取缺延胡索阴性样品3 g,同法制得阴性对照溶液,吸取上述供试品溶液、阴性对照溶液各20μL,对照品溶液10 μL,分别点于同一硅胶G薄层板上,以正己烷一氯仿一甲醇一二乙胺(l0:6:1:1)为展开剂,展开,取出,晾干,于碘蒸气中熏后,放置48 h,置紫外光灯(365 nm)下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的蓝色荧光斑点,阴性对照无相应斑点(图1 -G)。

-
+关注
私聊
-
yhl-87_
第36楼2009/11/06
薄层色谱法应用系列讲座(27)(下)-对乳安颗粒的质量控制
3 含量测定
3.1 色谱条件
色谱柱:大连Elite Hypersil ODS柱(200 mm×4.6 mm,5 μm);流动相:乙腈-0.05%磷酸溶液(15:85);流速1.0 mL·minˉ1,检测波长:230 nm;温度为室温。理论板数按芍药苷计算不低于4000。
3.2 溶液的配制
3.2.1 对照品溶液 精密称取经五氧化二磷减压干燥的芍药苷对照品适量,加50%甲醇制成每1 mL含0.052 mg的溶液。
3.2.2 供试品溶液 取本品1.0 g,精密称定,置50 mL具塞锥形瓶中,精密加入水饱和的正丁醇20mL,超声处理45 min,放冷,摇匀,滤过,精密量取续滤液10 mL置蒸发皿中,蒸干,残渣用50%甲醇溶解并定容至25 mL容量瓶中,用0.45 μm的微孔滤膜过滤,取续滤液作为供试品溶液。
3.3 线性关系考察与标准曲线的绘制 分别精密吸取浓度为52μg·mLˉ1的芍药苷对照品溶液1.5;2.5;5.0;7.5;10.0 mL,置5个10 mL量瓶中,加50%甲醇稀释置刻度,摇匀,各精密量取20μL注人高效液相色谱仪,记录色谱图,测定峰面积,以峰面积值(y)对进样量(X)进行回归,得回归方程为
y=1471317.7X+ 15845.7 r =0.9998
表明当芍药苷进样量在0.16~1.04μg之间时,其峰面积响应值与芍药苷的进样量有良好的线性关系。
3.4 精密度测定 分别精密吸取对照品溶液和供试品溶液各20μL,重复进样5次,对照品峰面积的RSD为0.16%,供试品峰面积的RSD为0.68%。
3.5 稳定性试验 取当日配制的供试液于0,4,8,12,16 h分别进样20μL,测得样品中芍药苷峰面积的RSD=1.51%,试验结果表明,样品供试液在16 h内稳定性良好。
3.6 回收率试验 取已知含量(芍药苷含量为0.13%)的同一批样品约0.5 g,共6份,精密称定,分为3组,每组分别加人154μg·mLˉ1的芍药苷对照品溶液3.0,4.0,5.0 mL,按供试品溶液制备项下方法制备,量取20μL进样,测得3个浓度的平均回收率分别为98.9%,101.6%,101.3%,RSD分别为1.5%,2.2%,3.6%。
3.7 重复性试验 按拟定的含量测定方法,对同一批样品分别制备样品试液5份,测得峰面积并计算含量,结果RSD=2.86%,重现性良好。
3.8 空白试验与供试品含量测定 精密称取不含白芍的阴性样品,按“供试品溶液的制备”项下操作,进样测定。结果表明:阴性样品在芍药苷出峰处无干扰,如图2所示。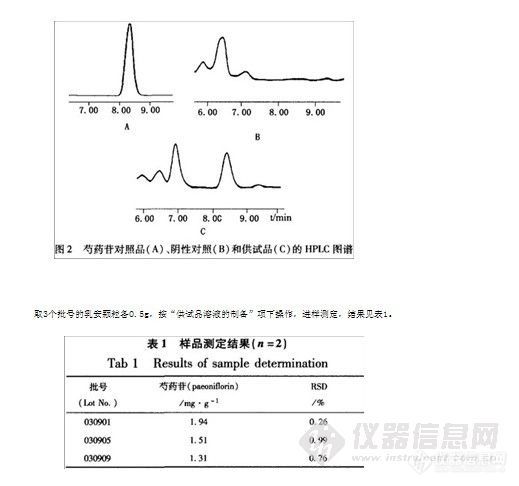
4 讨论
4.1 本实验对处方中七味药进行了定性鉴定,效果较好。其中柴胡、连翘采用先过柱后TLC鉴别,并改进了连翘TLC鉴别中的展开剂,斑点清晰,效果较好。茯苓的TLC鉴别供试液的制备参考了茯苓酸的提取方法,该方法简便、快速,效果较好。
4.2 中国药典用HPLC方法对多个样品进行了芍药苷的含量测定[4],本实验采用乙腈-0.05%磷酸为流动相,去除了盐,有利于保护色谱柱。本实验曾采用50%甲醇为提取溶剂,实验结果不甚理想,采用水饱和正丁醇为提取溶剂,避免了过柱操作,方法简便且提取率高杂质峰少,效果较好。高效液相色谱法测定乳安颗粒中芍药苷的含量,实验快速,结果准确、可靠,可作为乳安颗粒的质量控制分析方法。
参考文献
1 JI Yu - bin(季宇彬).Pharmacological Action and Application of Available Composition of Traditional Chinese Medicine(中药有效成分药理与应用)Heilongjiang(黑龙江):Heilongjiang Science and Technology Press(黑龙江科学技术出版社),1994.333
2 YANG Nan - song(杨南松),SUN Zhao—pu(孙照普),WANGYu -qing(王玉庆〉Studies on Quality Standard of HUANGJINGZANYU CAPSULE(黄精赞育胶囊质量标准的研究).Chin Tradit Herb Drugs(中草药),2002,33(9):798
3 JIN Chun—rong(新春荣),YOU Hui - Lian(尤慧莲),JIANG Zhi -ping(姜志萍).Study on quality standards for tiaoganhewei pills(调肝和胃丸质量标准的研究)Drug Stand China(中国药品标准),2003,4(6):41
4 CHP(中国药典).2000.Vo1 I(—部):233
-
+关注
私聊
-
yhl-87_
第37楼2009/11/06
薄层色谱法应用系列讲座(28)-测千金止咳丸中麻黄碱含量
薄层扫描法测定千金止咳丸中盐酸麻黄碱的含量
摘 要
目的 麻黄为千金止咳丸的君药,为确保本品质量,建立麻黄的含量测定方法.
方法 采用薄层扫描法测定盐酸麻黄碱含量,以氯仿、甲醇、浓氨试液(20:5:0.5)为展开剂,1%茚三酮溶液为显色剂;双波长反射法锯齿扫描:λs = 510nm λR = 640nm.
结果 通过方法学考察,点样量在1.454~14.54μg 范围内呈良好的线性关系回归方程为Y=3830.48X+6229.85, r=0.997 平均回收率为99.4% n=6 .
结论 试验表明,该方法可靠,数据准确,操作简便易行.
本文由成都中医药大学的老师们 夏厚林、万丽、赵勇智、范成杰等共同完成,现全文介绍如下
前 言
千金止咳丸具有宣肺化痰、止咳平喘之功效,用于外感风寒咳嗽、痰热内蕴等症。 麻黄为方中君药,其主要有效成分为生物碱。 根据文献报道,麻黄生物碱的含量测定有薄层扫描法、HPLC 法、GC 法、非水滴定法 [1-4]等,因本品为多味中药的大复方,其余药味对麻黄的测定有干扰,现采用薄层扫描测定对本品麻黄中所含生物碱类成分进行含量测定研究.。
实 验
1 仪器与试药
CS- 930 型双波长薄层扫描仪(日本岛津);自动点样仪(瑞士CAMAG 公司);硅胶G (青岛海洋化工);千金止咳丸(自制);盐酸麻黄碱对照品(中国药品生物制品检定所,供含量测定);试剂均为分析纯。
2 实验部分
2.1 对照品溶液的制备
精密称取经干燥至恒重的盐酸麻黄碱对照品7.27mg, 置5ml 量瓶中用少量甲醇溶解后加甲醇至刻度(1.454mg/ml), 摇匀,即得.
2.2 供试品溶液的制备
取本品,研细,精密称取粉末约5g, 置100ml 量瓶中,加甲醇适量(近刻度),超声处理(250W 50KHz) 1 小时,放冷,加甲醇置刻度,摇匀,滤过,弃去初滤液,精密量取续滤液60ml, 置蒸发皿中,水浴上蒸至近干,残渣加甲醇使溶解,定容至5ml 量瓶中,作为供试品溶液。
2.3 色谱条件
薄层板:硅胶G 板(厚约0.5mm), 105℃ 活化40min; 展开剂:氯仿-甲醇-浓氨试液(20:5:0.5); 显色方式:薄层板展开后晾干,于80℃ 烘箱烘20min, 放冷,再喷以1%茚三酮溶液,105℃烘至斑点清晰,立即覆盖相同大小的玻璃板,周围用胶布固定。.
2.4 扫描条件
双波长反射法锯齿扫描: λs = 510nm,λR = 640nm ;狭缝:0.4mm ×0.4mm ;灵敏度:中;线性化参数:SX=3.
2.5 稳定性考察
对同一薄层板的同一斑点依法测定,每隔20min 测定1 次,结果表明,斑点在3h 内稳定,RSD 为2.8% (n=9) .
2.6 线性关系考察
分别精密吸取对照品溶液1、 2 、4、 6 、8、 10 μl 点于同一硅胶G 薄层板上,依法展开,显色,扫描测定.。 以点样量(X,μ g) 为横坐标,斑点面积积分值(A )为纵坐标绘制标准曲线。 结果表明,盐酸麻黄碱点样量在1.5~15.0μ g 范围内点样量与斑点面积积分值呈线性关系,其回归方程A=3830.48X+6229.85 r=0.997, 由于直线不通过原点因而采用外标两点法。
2.7 精密度考察
精密吸取对照品溶液2μl ,在5 个薄层板上分别点样5 次,依法全部展开,显色,扫描测定。 其同板精密度RSD 为1.5% n=5 , 异板精密度RSD 为4.3%。
2.8 重复性试验
精密称取同一批次样品5 份,按拟定的含量测定方法测定盐酸麻黄碱的含量,结果RSD 为4.1%.
2.9 回收率试验
精密称取已知含量2.170mg/g 的同一批号样品,按样品中盐酸麻黄碱含量的40% 、50%及60%分别精密加入盐酸麻黄碱对照品,依法提取测定., 平均回收率为99.4% ( n=6) RSD 为4.0%.。
2.10 干扰因素的考察
取缺麻黄的千金止咳丸自制样,按供试品溶液制备方法制成阴性对照液,依法展开显色扫描测定., 结果在盐酸麻黄碱对照品相应的色谱位置无吸收,表明其它成分对盐酸麻黄碱的测定无干扰.。
2.11 样品测定
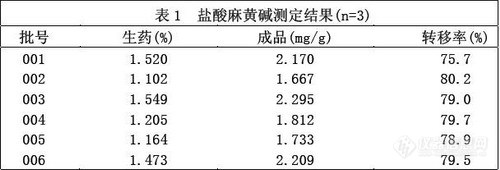
精密吸取供试品溶液2 μl 对照品溶液1 μl 、2μ l 分别交叉点于同一硅胶G 薄层板上,依法展开,显色,扫描侧定.。结果见表1.。
2.12 麻黄药材中盐酸麻黄碱的含量测定
精密称取麻黄药材粉末1.0g ,依法提取测定., 结果见表1.
3 讨论
(1) 千金止咳丸为部标第七册收载的中成药品种,本文为建立该品种质量标准含量测定项提供了依据.
(2) 生药与成品间的转移率达到75%以上,说明了制备工艺的合理性.。
(3) 薄层色谱法测定麻黄碱含量时,提取时间是重要影响因素,故测定时间应严格控制。
(4) 提取条件考察中,对样品浸泡时间考察(过夜)表明,浸泡时间对样品含量无明显影响。
参考文献:
[1] 罗远秀. 灵仙化石胶囊质量标准的研究 [J]. 广西医学2000 ,22 (2) 124-128.
[2] 陈发奎, 郭允珍, 鹿野美弘. 用高效液相色谱法同时测定九分散中麻黄碱和士的宁的量[J]. 中草药1994 ,25 (11) 271-275.
[3] 常珉, 王婉钢, 张立群, 等. 气相色谱法简易测定五种中成药中麻黄碱的含量 [J]. 中成药1989, 11 (2): 11-14.
[4] 卫生部药典会编. 中华人民共和国药典. 1995 年版一部, 1995.
-
+关注
私聊
-
yhl-87_
第38楼2009/11/06
薄层色谱法应用系列讲座(29)-加压薄层色谱的原理和应用
加压薄层色谱法的原理及其应用
摘 要:对加压薄层色谱的原理和应用作以介绍。介绍了加压薄层色谱的仪器结构、工作方式;对影响加压薄层色谱的因素、常见问题及解决方法进行了总结;简要地介绍了该方法在分析中的应用情况。
本文由中国药品生物制品检定所的何 轶、鲁 静、林瑞超三位研究人员共同完成,对加压薄层色谱法的原理和应用有详细介绍,值得参考,现全文介绍如下
前 言
薄层色谱法作为一种快速简便的色谱技术已在中草药分析、毒物分析等众多领域中广泛应用。在传统薄层色谱法中,展开剂依靠毛细作用通过薄层吸附剂来完成对组分的分离,因此其分离时间不可控,而且随着展开距离的增加,溶剂前沿的移动逐渐减慢,被分离组分的扩散也越来越严重。针对此情况,分析学家对薄层系统进行了改进,发展了薄层色谱的一个重要分支——强迫流动薄层色谱(forced¨flow planar chromatography,FFPC)。 FFPC是通过外力强迫展开剂在吸附剂中运动,它主要有离心薄层色谱法(rotation planar chromatography,RPC)与加压薄层色谱法(overpressured-layerchromatography,0PLC)两种。RPC通过高速旋转产生的离心力加速展开剂的运动;OPLC则依靠加压泵将展开剂直接泵入薄层板中,并通过泵来调节展开剂的流速[1,2]。
20世纪60年代出现了薄层色谱超微展开室。这种展开室在展开过程中用一块玻璃或塑料膜覆盖在薄层板的表面,从而减少了薄层分离中气相的影响。1979年Tyihak在超微展开室的基础上,在覆盖薄层板的塑料膜上加以一定的压力,并将展开剂压入薄层板,从而发展了OPLC技术;在此之后,该方法在理论、分析条件及应用等方面的研究得到了不断的深人,商品化的OPLC仪及其专用的薄层板也不断推出[1]。本文以市售的OPLC仪为例,对OPLC进行介绍。
实 验
1 仪器结构
薄层板夹(cassette)是0PLC技术的关键部分。板夹主要由两层构成,上层为聚四氟乙烯薄膜层,下层为一块钢板,薄层板吸附剂面向上置于聚四氟乙烯薄膜与钢板之间。在聚四氟乙烯薄膜两边均有一个小孔,溶剂通过这两个小孔进出薄层板。在孔的两侧与薄层板接触的一面刻有凹槽,使溶剂可以快速到达薄层板边缘,从而保证薄层板中间和边缘几乎同时开始展开。在薄层板的边缘有一圈约2mm宽的吸附剂被刮掉,再用高分子材料加上一层密封条,以防止加压展开时展开剂从薄层板边缘溢出。板夹下层的钢板主要起支撑作用。薄层板夹的工作原理示意图见图1。
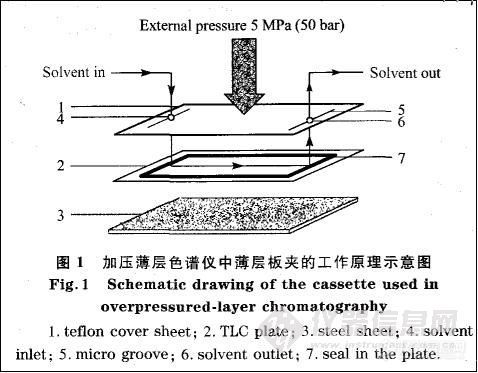
工作时用一个泵在板夹上加压,最大压力可达5 MPa(50 bar),同时用另一个泵将展开剂输人到薄层板中,以完成分离过程门。OPLC中常用的薄层板有5 cm×20 cm、10 cm×⒛cm以及⒛cm×⒛cm等规格,不同大小的薄层板使用不同的板夹。
OPLC仪主要由两部分构成,其结构如图2所示。仪器上层主要有电源、控制系统以及泵系统等;下层为展开室,工作时将板夹插入。仪器具有独立的加压泵和输液泵系统,可进行分析和制备等工作,并可通过阀切换实现简单的梯度洗脱。此外还有主要针对半制各工作设计的OPLC仪,它仅有一个加压泵和一个展开室,展开剂的输送可通过连接HPLC泵来实现,因此构成一台OPLC仪的成本也较低。
2 工作方式
OPLC属于薄层色谱的一个分支,但由于它采用了泵输送溶剂,因此可进行类似于高效液相色谱的在线分析,也因此有多种工作方式可供选择[3]。
2.1 离线点样—分离-离线扫描
这种工作方式类似于传统的薄层色谱。首先将样品点于薄层色谱板上,然后进行分离,分离后取出薄层板进行定性定量分析。在这种方法中可以同时进行多个样品的分析,此外也可选择多种显色剂进行显色,以提高分析的专属性和灵敏度。
2.2 离线点样-分离—在线检测
在OPLC仪后可以串联高效液相色谱用的紫外或其他类型的检测器,从而进行在线检测。可将样品点于薄层色谱板上用展开剂将其连续洗脱后用检测器测定洗脱液。
2.3 在线进样—分离—离线扫描
在展开室前可以连接高效液相色谱用手动进样器,将薄层板用展开剂充分冲洗润湿后直接在线进样,样品分离后取出薄层板进行检测。这种工作方式每次只能分析一个样品。
2.4 在线进样ˉ分离ˉ在线检测
样品经在线进样分离后也可串联高效液相色谱用检测器进行在线检测。这种方法和高效液相色谱较为相似。因此也有人将OPLC称为平面柱色谱。
针对不同的实验要求可以选择不同的分析方式,这是OPLC一个很突出的优点。

-
+关注
私聊
-
yhl-87_
第39楼2009/11/06
薄层色谱法应用系列讲座(29)(下)-加压薄层色谱的原理和应用
3 加压薄层色谱的分离效果及其影响因素
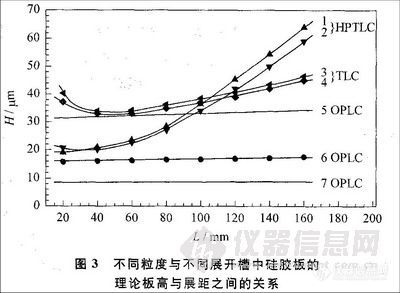
一般来说,OPLC的分离效果较传统薄层色谱好,原因主要有以下几方面。首先,正如文章开头所提到的,OPLC通过泵将展开剂输送至薄层板,使分析时间缩短,待分离组分的扩散也较小。这一现象在展距增加的情况下更为明显。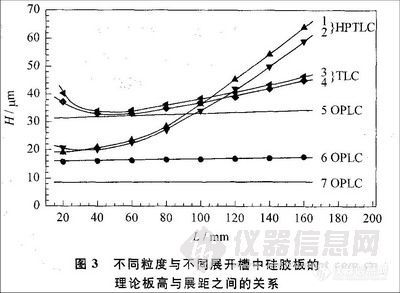
从图3中可以看出,随着展开距离的增大,传统薄层色谱中的理论板高迅速增大,但是在OPLC中理论板高变化不大L[,3」。所以在OPLC中可通过增加展距来达到提高分离效果的目的,也因此在OPLC中薄层板的长度多为⒛cm。此外,在传统薄层色谱中,当展开剂到达薄层板的边缘后就不能再进行分离了。但在OPLC中可以用泵连续输送展开剂,展开剂溢出薄层板后分离仍可继续进行,从而提高了比移值较小组分的分离度。这些都对提高分离效果起到了重要作用。
影响OPLC分离的因素主要有以下几方面。首先,OPLC展开室中空间较小,故气相对OPLC分离的影响较少,也有利于提高OPLC的重现性。但是在薄层板的吸附剂颗粒的间隙中仍然存在少量气体,它们会对分离结果产生一定的影响。离线点样与在线进样的分离结果会有一定的差别,就是因为在在线进样前已经充分冲洗润湿了薄层板,去除了其中的气体。此外,吸附在吸附剂表面的气体也会对分离的结果产生影响,在“4.2”节中专门对此进行介绍。
与高效液相色谱相似,OPLC中展开剂的流速也会对分离结果产生影响。流速过大和过小均会导致分离度降低[1]。在分析过程中,输液泵的压力应不大于加压泵的压力,否则展开剂可能从板夹中溢出。在目前市售的OPLC仪中,当输液泵压力达到加压泵压力的80%时,系统报警并自动停止工作。
在板夹上所加压力的大小也会影响到分离效果。文献[1]表明在采用离线工作方式时,所加压力越大,理论板高越小;但是在在线工作方式中,板夹上所加的压力对理论板高影响不大。一般在分析过程中选择使用仪器所能提供的最大压力。早期的0PLC仪提供的最大压力为2,5 MPa(25 bar),现在的OPLC仪最大可提供5 MPa(50 bar)的压力。
此外,温度、所使用的展开剂以及吸附剂的粒度等也会对分离效果有一定的影响,这些影响和传统的薄层色谱相似,不再——讨论。
4 加压薄层色谱中的常见问题及解决方法
4.1 多溶剂前沿(multiˉfront)问题
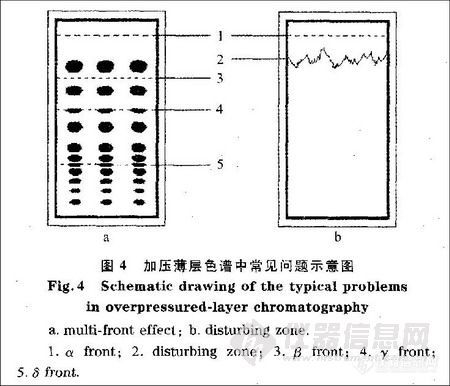
在传统的薄层色谱中,如果使用多种溶剂的混合溶液作为展开剂,并且展开前没有充分地预饱和,展开剂的不同组分就可能在薄层板上发生分离,从而出现多个溶剂前沿。在离线工作方式的加压薄层色谱中由于减少了气相对色谱的影响,这一现象较传统薄层色谱更加严重。位于第二或其他溶剂前沿的组分通常呈窄条状9如图4-a所示。
如果同时有多个组分位于此处,则很难将其分离开。但是如果在进行制备时,待分离组分位于第二或第三溶剂前沿位置并无其他组分干扰,则只用很小体积的展开剂就可以将该组分洗脱下来。在使用同一个展开剂时,多溶剂前沿出现的位置是固定的,因此可以用同一个样品在距展开起始位置不同的高度多次点样,观察这些样品点在不同位置的峰形变化来判断溶剂前沿的位置[3’4]。可以通过改变溶剂的配比来调节多溶剂前沿的位置。如果仍不能满足要求,则需更换所使用溶剂的种类。
4.2 干扰带(disturbing zone)问题[3,5]
在离线工作方式的OPLC中仍有一部分气体残留。这些气体大部分存在于硅胶颗粒的间隙中,同时还可吸附于硅胶表面。这些气体是出现干扰带的主要原因。在展开过程中,溶剂穿过硅胶颗粒的间隙,将游离的气体推出。同时,吸附剂吸附一部分展开剂分子并释放原来吸附的气体分子。但是,当解吸附的速度比展开剂移动速度低时,被吸附的气体就不能随着游离气体一起被推出而是释放于展开剂中。当展开剂中溶解的气体超过其饱和浓度时,气体分子就会形成微小的气泡c这样在已经饱和的展开剂和未饱和的展开剂之间就可以观察到干扰带。因为含有饱和和未饱和气体的展开剂的折射率不同,因此可以清楚地用肉眼观察到薄层板上的干扰带。从理论上说,干扰带应呈一条直线,但是由于受吸附剂粒度不均匀的影响,在实验中所观察到的干扰带多呈锯齿状,如图4-b所示。
由于干扰带是因被吸附的气体不能被快速地释放所产生,因此可以通过调节展开剂的流动速度(即推动展开剂移动的压力大小)来调节干扰带的位置。一般说来,对于同一种吸附剂和展开剂,在干扰带出现的位置上,流速不同的展开剂所受的压力是相同的,当展开剂的流动速度增大时,推动展开剂移动的压力也增大,被吸附的气体就容易从薄层板上解吸,干扰带向前移动。此外还可以通过改变溶剂种类,增加展开剂对气体的溶解度来避免干扰带的产生。如果以上两个办法均不能去除干扰带的影响,还可在展开前用不推动样品移动的溶剂预洗薄层板。如欲分离极性化合物时用石油醚等非极性溶剂预洗硅胶薄层板,使被吸附的气体充分释放后再进行展开,以减少残存气体的干扰。
以上问题均在离线工作方式的加压薄层色谱中存在,在在线工作方式中,因为已经将薄层板充分润洗,这些问题均不会发生。
5 应用 i
从20世纪60年代出现首台0PLC仪以来,OPLC在植物药分离制备、尿样分析等多领域均得到了广泛的应用。下面举例简要说明其应用情况。
Dallenbach-Toelke等[6]采用普通薄层色谱、高效薄层色谱(IIPTLC)、0PLC和超微室离心薄层色谱(ultra micro- chamber centrifugal layer chromatography,UCLC)对药用婆婆纳Veronica officinalis中Szikszay等[7]用Automatic Personal OPLCBS-50加压薄层色谱仪(OPLC-NIT Ltd)对phthaloyl-amlodi pine的纯度进行了测定。
在高效薄层板上,以正己烷-醋酸丁酯-醋酸乙酯-氯仿(体积比为60∶15:15:20)为展开剂,薄层板压力5 MPa(50 bar),展开剂体积8 500 pL,所测定的有关物质在0.05~2.00 pg内线性关系良好,测定结果的相对标准偏差(RSD)为2.9%。
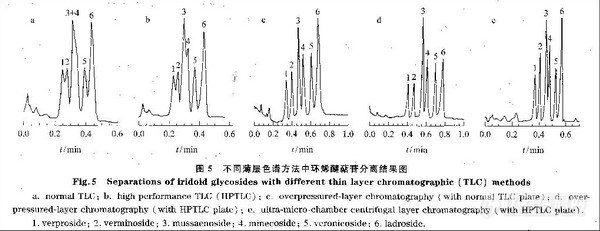
Mincsovics等r:]利用OPLC对菊科植物Leuzea提取物中的葫芦巴碱(trigonelline)进行了制各。他们将Leuzea的甲醇提取物点于OPLC硅胶薄层板上,先用展开剂(正丙醇¨甲醇-水(体积比为40:6:60))从出口向人口方向预洗薄层板,然后再从人口到出口方向输送展开剂进行分离。预洗不仅可去除薄层板上的气泡,减少干扰带的产生,同时还可去除样品中部分较易洗脱的植物色素,减少它们对待分离组分的干扰。此外,他们的研究表明,OPLC很适合进行微量制各,一次可对2~5 mg的样品进行分离,得到0,1~0.5 mg的纯品。据介绍,的环烯醚萜苷类化合物进行了分离。展开剂均为醋酸一丁酮-氯仿-水(体积比为25:j/⒌37.5:7.5)。目标化合物在普通薄层色谱中的展距为9 cm;在HPTLC中的展距为4.5 cm;在OPLC和UCLC中的展距均为17 cm(OPLC仪为Chrompres-10(LaborMIM,Budapest,Hungary),薄层板压力为1 MPa(lObar),输液泵压力为0,15 MPa(1.5 bar),先用丁酮对薄层板进行预洗,以去除吸附剂吸附的气体,减少干扰带的影响;UCLC为该实验室自制,采用高效薄层板进行分离)。各种薄层色谱的分离结果见图5,可以看出,普通薄层色谱不能将玉叶金花苷(mus ̄saenoside)和米内苷(minecoside)分开,HPTLC仅能将两者刚刚分开。如果在这两种薄层色谱中再增加展开距离,则均导致样品的扩散加剧,因此作者采用FFPC对该样品进行了分离。0PLC和UCLC均可获得较好的分离效果,其中以OPLC效果最好。加压薄层色谱仪还可用专门的板夹使用制备薄层板进行制备,从而使一次制备量达到几十毫克。
-
+关注
私聊
-
yhl-87_
第40楼2009/11/06
薄层色谱法应用系列讲座(30)一种新的展开剂用于薄层色谱分析
摘 要:以十六烷基三甲基溴化铵(CTAB)/正丁醇/正辛烷/水微乳液作为一种新的展开剂,对23种氨基酸进行了薄层色谱分析。研究了微乳液含水量和氨基酸分子结构对Rf
值的影响;同时选择含水量为40%的微乳液对氨基酸混合物进行了分离鉴定并与传统的展开剂作了比较;此外还探讨了微乳色谱理论。研究结果表明,当微乳液为油包水型及油水双连续型时,氨基酸的Rf值多在0.2与0.8之间,色谱分析结果令人满意。
本文由湖北民族学院化工系的田大听、史伯安和谢艳等老师们共同完成的,氨基酸的液相色谱和离子色谱法已经比较成熟,而在薄层色谱法上有自己分析观点的文章不多,可惜没提供TLC分析原图。现全文介绍如下。
(
前 言
微乳液是由水、油、表面活性剂及助表面活性剂等在一定配比下自然形成的、无色透明的、低粘度的热力学稳定体系。由于它具有独特的结构与性能,因而备受人们的关注,现已广泛应用于三次采油、化妆品、药剂学、润滑、酶催化、有机合成及无机合成等领域[1~3]。近年来,已经有文献报道将其作为展开剂用于生物碱[4]和氨基酸[5]的薄层色谱(TLC)分析。作者所在课题组曾用十二烷基硫酸钠/正丁醇/正己烷/水微乳体系对氨基酸进行薄层色谱研究[5],所用表面活性剂是阴离子型。本文选用十六烷基三甲基溴化铵(CTAB)这种阳离子型表面活性剂,并以CTAB/正丁醇/正辛烷/水微乳液作为一种新的展开剂,对氨基酸进行薄层色谱分析,以进一步搞清其中的有关规律,并进一步拓宽微乳液的应用领域。
1 实验部分
1.1 试剂与仪器
十六烷基三甲基溴化铵(CTAB)、正丁醇、正辛烷、茚三酮均为分析纯;23种氨基酸均为色谱纯(除DL-β-苯丙氨酸外,其余皆为L型),用二次蒸馏水配成1g/L溶液。硅胶G薄层板,展开池。
1.2 微乳液的配制
本实验所用表面活性剂为CTAB,助表面活性剂为正丁醇,油为正辛烷,水为二次蒸馏水。在保证前三者质量比一定的条件下,改变水的含量,配制成系列微乳液。
1.3 实验方法
用毛细管吸取少量氨基酸溶液于硅胶G板上点样,以上述各微乳液为展开剂,用上行法展开,待展开到一定距离后,取出晾干。用茚三酮显色剂显色,然后计算比移值Rf
2 结果与讨论
2.1 实验结果
微乳液的成分配比及结构见表1。
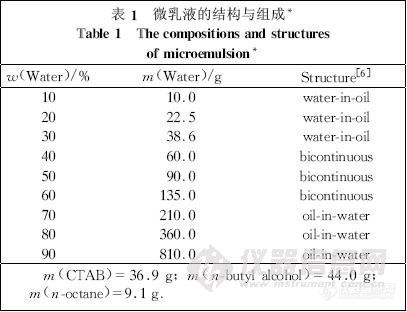
从CTAB、正丁醇、正辛烷和水体系的拟三元相图[6]可知,在固定CTAB、正丁醇和正辛烷适当质量比的情况下,改变水的含量(质量分数),可得到一个从油包水区(water-in-oil,W/O区)到油水双连续区(bicontinuous,BC区)进而到水包油区(oil-in-water,O/W 区)的连续单相微乳区。以据此所配的系列微乳液为展开剂,23种氨基酸的薄层色谱结果见表2。
2.2 微乳色谱理论探讨
在微乳色谱中,溶质在固定相、油或水连续相及内核和界面等各相之间进行分配。色谱分析过程中,氨基酸分配在微乳液的内相或连续相处,也可穿插在界面上由CTAB与正丁醇组成的栅栏层中,另外吸附剂硅胶G也对其产生吸附作用,使其在与富集相的竞争中连续不断地脱附出来。由于吸附、分配、静电、疏水、立体、萃取与反萃取等可能的效应,导致各氨基酸迁移速率不同而使Rf值有所差异。
2.3 微乳液含水量对Rf值的影响
从表2可以看出,样品的Rf值随着微乳体系含水量的增加而增大。在薄层色谱分析中,Rf值主要受样品的极性、吸附剂的活度及展开剂的极性这三者的影响。当前二者恒定时,其Rf值主要受展开剂的极性控制。一般来说,展开剂极性愈大,Rf值也愈大。通过调节含水量,可获得适宜的展开体系。而微乳液含水量过高(大于60%),会使体系极性过大,导致许多斑点迁移至前沿线(湿线)边沿,且拖层严重,多数斑点形状不规则,可见微乳液的含水量以10%~60%为宜。在W/O型微乳液中,游离氨基酸进入水相(内核),得到富集增溶。在油水双连续相中,水既是增容相,又是连续相,游离氨基酸在两相均被适当富集;由于微乳液特殊的热力学稳定作用,使得富集氨基酸受多种效应影响,其中特定结构的氨基酸以特定速率被释放出来,从而得到较好的色谱效果。但如果水量过大,即O/W 型微乳液中,氨基酸主要进入连续相,这样微乳液的特点无法表现出来,此时则与普通极性展开剂无异。
2.4 氨基酸分子结构对Rf值的影响
样品极性越大,则被吸附剂吸附得越牢固,在相同条件下,迁移速率就越小。在主要结构相同(如表2中6#与2#,18#与17#氨基酸等)的条件下,分子中含OH 比不含OH 的氨基酸的Rf值要小。
值得一提的是,表2中15#与6#,17#与18#,20#与21#,15#与23#,它们的Rf值有些特殊,其值不仅与分子结构有关,而且还与微乳液的含水量有关。当含水量小于30%时,Rf(15#)>Rf(6#),Rf(17#)>Rf(18#),Rf(20#)>Rf(21#),Rf
(15#)>Rf ( 23#);当含水量大于30%时,则刚好相反。6# 与15# 相比,前者含OH ,后者含SH ;17#与18#相比,20#与21#相比,后者比前者多OH ;而15#与23#相比,后者是前者的盐酸盐。总之,从分子结构上看,这些样品中后者的极性比前者强。一方面,当展开剂极性较低时,如含水量小于30%时,Rf值受分子极性控制;另一方面,当微乳液含水量较大时(大于30%),此时微乳液开始由W/O型向油水双连续型及O/W型转化,则Rf值不仅受分子极性、展开剂极性的影响,而且受静电、疏水、立体、萃取与反萃取等因素的影响,也可说随微乳液类型的转化,有的样品的Rf值变化大,有的变化小。
2.5 本法对氨基酸混合物的应用及与不同类展开剂的比较
对几种氨基酸的混合物,选择含水量40%的微乳液作展开剂,用同种氨基酸作对照,点样,展开结果表明其分离效果明显。
另外,按传统方法,采用CH3CH2OH,H2O与CH3COOH的混合液(体积比为50:50:1)为展开剂,与本法所用展开剂进行比较。结果表明前者展开后斑点大多不规则,且易产生拖尾现象,而本法则很少有这种情况。这说明,正是由于微乳液具有独特的结构,因此本法所用展开剂对目标分析物的色谱性能良好。