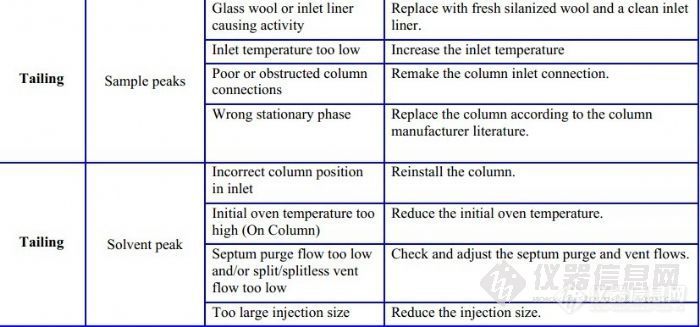
其实我也在怀疑衬管的情况,难道之前用乙醇超声把某些活性位点激活了,而且我们这里没有硅烷化试剂做灭活处理,刚找到一个所谓的新衬管,里面全是石英棉的毛,正用甲醇泡着,估计泡不干净,又不敢再去超声了,但是师姐说她们那里就是超声清洗的,哎,不知道该听谁的
tdp001(tdp001) 发表:你没有提到你测定样品性质,所以只能给你提一下建议:
1、不用轻易切色谱柱;
2、不加内标物,进微量样品测试一下,看峰形如何,我有些担心色谱柱;
3、有些担心衬管的清洗问题,担心残留影响分离效果。你换一根新的衬管看看,如果好了,就是清洗方法有些不妥。