推荐厂家
暂无
暂无
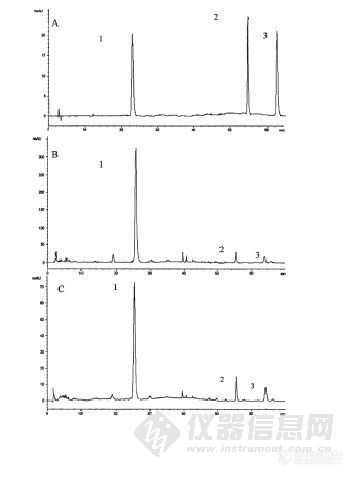
作者:王洋(南京中医药大学)摘要:本文对不同采收期、不同贮存时间的广陈皮药材主要成分的含量进行了测定,归纳总结了其主要的变化规律,旨在揭示个青皮与广陈皮药材“同源不同性”的机理以及初步探索贮存时间对广陈皮内在成分的影响,为广陈皮药材的临床应用提供实验依据。 1.本文所需广陈皮药材样品共13份,收集于广东省新会县广陈皮种植基地,样品分别为2004年11月、2005年10月、2005年11月、2005年12月、2006年10月、2006年11月、2006年12月、2007年5/6月、2007年8月、2007年9月、2007年10月、2007年11月和2007年12月。 2.对各广陈皮样品的主要成分进行了含量测定,确定了测定方法。总黄酮、总生物碱和总多糖采用紫外分光光度法,回归方程、相关系数和线性范围分别为:y=3.397x+0.011,r=0.9998,线性范围:0.04mg/m1~0.24mg/ml;y=7.12x+0.018,r=0.9998,线性范围:0~0.18mg/ml;y=0.083x-0.005,r=0.9997。挥发油成分采用GC/MS法测定,气相色谱条件:色谱柱为DB-5(30m×250μm×0.25μm)石英毛细管色谱柱;进样口温度220℃;程序升温60℃(维持5min),以5℃/min升温至200℃;载气为高纯氦气,流量1mL/min,溶剂延迟3min。质谱条件:MSD离子源为El源,离子源温度230℃,电子能量70eV,扫描质量范围50~550质量数。加速电压1000eV。 橙皮苷、川陈皮素和橘皮素的含量测定采用HPLC法,色谱条件为:流动相体系为甲醇(A)-乙腈(B)-4%醋酸水溶液(C),梯度洗脱。0~25min,A: B:C=10:15:75;25~40nun,A升至50%,A:B:C=50:15:35,保持30min。色谱柱为Platisil ODS柱(5μm,250×4.6mm),柱温30℃,流速1ml/min。检测波长:0~50min,283nm;50~70min,332nm。 3.对不同采收期广陈皮药材主要成分的含量变化进行了对比,结果为:个青皮总黄酮和橙皮苷的含量最高,而广陈皮药材挥发油、生物碱以及多糖类成分均高于个青皮,这可能就是个青皮与广陈皮“同源不同性”的内在机理。 对于不同贮存时间的广陈皮药材,黄酮类成分与总生物碱含量随着贮存时间的延长会基本保持不变或者略有上升。 4.用“老化”模拟实验处理广陈皮药材,结果表明在高温条件下,黄酮类成分化学性质较稳定,且随着烘烤时间的延长,含量会略有上升。另外,在高温条件下,药材内部可能发生了化学成分的转化并产生新的极性较大的物质。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208131431_383497_1606903_3.jpg
各位版友:大家好!我最近在做一个关于中药陈皮的制剂,现在在做质量标准。我打算做一个:陈皮对照药材,橙皮苷,我的制剂样品。我按照药典陈皮的方法来做, (2)取陈皮对照药材粉末O.3g,加甲醇10ml,加热回流20分钟,滤过,取滤液5ml,浓缩至1ml,作为供试品溶液。另取橙皮苷对照品,加甲醇制成饱和溶液,作为对照品溶液。照《薄层色谱法检验标准操作程序》(附录ⅥB)试验,吸取上述两种溶液各2μl,分别点于同一用0.5%氢氧化钠溶液制备的硅胶G薄层板上,以乙酸乙酯甲醇一水(100:17:13)为展开剂,展至约:3cm,取出.晾干,再以甲苯一乙酸乙酯——甲酸一水(20:10:1:1)的上层溶液为展开剂,展至约8cm,取出,晾干,喷以三氯化铝试液,置紫外光灯(365nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。但是发现供试品出不来斑点,知道为什么吗?各位大侠,知道应该怎么做吗,请指点一下。橙皮苷也很难显色,有谁知道为什么吗?浓度已经很大了。
本人最近在用薄层鉴别陈皮药材,发现药典的规定是:吸取上述对照品和对照药材及供试品,分别点于同一用0.5%氢氧化钠溶液制备的硅胶G薄层板上,以乙酸乙酯甲醇一水(100:17:13)为展开剂,展至约:3cm,取出.晾干,再以甲苯一乙酸乙酯甲酸一水(20:10:1:1)的上层溶液为展开剂,展至约8cm,取出,晾干,喷以三氯化铝试液,置紫外光灯(365nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的荧光斑点。我按照上述方法进行操作,有几个疑问:薄层检测陈皮药材时,为什么不用CMC-Na铺板?而用NaOH呢?难道他们会反应?但用NaOH铺的板太不结实了。展开系统又是如何摸索出来的?两次展开,目的何在?刚刚接触薄层色谱,有点不明白。请高手指教。







 400-860-5168转4139
400-860-5168转4139
 留言咨询
留言咨询
 400-860-5168转3905
留言咨询
400-860-5168转3905
留言咨询
 留言咨询
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP
