推荐厂家
暂无
暂无
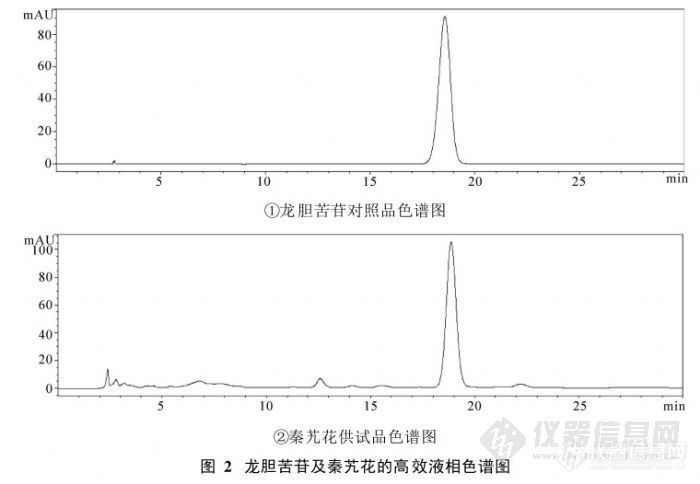
【作者】:胡一晨, 万丽, 张艺, 杨芳, 吉琅, 廖晴, 曹纪亮【摘要】: 目的: 建立藏 药秦艽花的质量标准。方法: 运用薄层色谱法 对秦艽花进 行定性鉴别 ; 采用 高效液相 色谱法测定秦艽花中龙胆苦苷的含量, 色谱柱为 D IKM A D iam onsil C18 ( 250 @ 4. 60 mm, 5 Lm ) 柱, 流动 相为甲醇 - 水 ( 25: 75), 检测波长为 275 nm, 流 速 1. 0 mL# m in- 1, 柱温 30 e 。结果: 薄层 鉴别斑点清 晰, 重 复性好; 高效液 相色谱法测 定龙胆苦苷 在 0. 912Lg~ 4. 560 Lg 之间呈良好的线性关系, 平 均加样回收率为 98. 62% , R SD 为 1. 21% 。结论: 该方法简单可行, 重复性好, 可作为藏药秦艽花的质量控制方法。【作者单位】: 成都中医药大学药学院 中药材标准化教育部重点实验室 中药资源系统研究与开发利用省部共建国家重点实验室培育基地【关键词】: 秦艽花; 龙胆苦苷; 质量标准http://ng1.17img.cn/bbsfiles/images/2012/07/201207311325_380846_1838299_3.jpg
胡黄连、小秦艽花的薄层怎么做?用3个不同的展开剂?
[size=20px][color=#93c6bc][b]鉴别[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [font=宋体][/font][b][font=宋体][/font][/b] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [b][font=宋体][/font][/b][font=宋体](1)取本品粉末0.5g,加甲醇10ml,超声处理15分钟,滤过,取滤液作为供试品溶液。另取[color=var(--weui-LINK)]龙胆苦苷[i][/i][/color]对照品,加甲醇制成每1ml含1mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取供试品溶液5μl、对照品溶液1μl,分别点于同一硅胶GF[sub]254[/sub]薄层板上,以乙酸乙酯-甲醇-水(10:2:1)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。[/font] [font=宋体](2)取栎瘿酸对照品,加三氯甲烷制成每1ml含0.5mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取〔鉴别〕(1)项下的供试品溶液5μl和上述对照品溶液1μl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-甲酸(50:1:0.5)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点。[/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font] [font=宋体][/font][font=宋体][/font] [font=宋体] [/font] [size=20px][color=#93c6bc][b]检查[/b][/color][/size][size=16px][color=#e2a4a4]|[/color][/size] [font=宋体][/font] [b][font=宋体][/font] [font=宋体][/font] [font=宋体]水分 [/font][/b][font=宋体]不得过9.0%(通则0832第二法)。[/font] [b][font=宋体]总灰分[/font][/b][font=宋体] [/font][font=宋体]不得过8.0%(通则2302)。[/font] [b][font=宋体]酸不溶性灰分 [/font][/b][font=宋体]不得过3.0%(通则2302)。[/font] [b][font=宋体]【[color=var(--weui-LINK)]浸出物[i][/i][/color]】[/font][/b][font=宋体] 照醇溶性浸出物测定法(通则2201)项下的热浸法测定,用乙醇作溶剂,不得少于24.0%。[/font] [b][font=宋体]【含量测定】[/font][/b][font=宋体] 照高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法(通则0512)测定。[/font] [b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] [/font][font=宋体]以[color=var(--weui-LINK)]十八烷基硅烷键合硅胶[i][/i][/color]为填充剂;以乙腈-0.1%醋酸溶液(9:91)为流动相;检测波长为254nm。理论板数按龙胆苦苷峰计算应不低于3000。[/font] [b][font=宋体]对照溶液的制备[/font][/b][font=宋体] [/font][font=宋体]取龙胆苦苷对照品、马钱苷酸对照品适量,精密称定,加甲醇分别制成每1ml含龙胆苦苷0.5mg、马钱苷酸0.3mg的溶液,即得。[/font] [b][font=宋体]供试品溶液的制备 [/font][/b][font=宋体]取本品粉末(过三号筛)约0.5g,精密称定,置具塞锥形瓶中,精密加入甲醇20ml,超声处理(功率500W,频率40kHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。[/font] [b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]分别精密吸取两种对照品溶液与供试品溶液各5[/font][font=&]~[/font][font=宋体]10[/font][font=宋体]μl,注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url],测定,即得。[/font] [font=宋体]本品按干燥品计算,含龙胆苦苷(C[sub]16[/sub]H[sub]20[/sub]O[sub]9[/sub])和马钱苷酸(C[sub]16[/sub]H[sub]24[/sub]O[sub]10[/sub])的总量不得少于2.5%。[/font]







 留言咨询
留言咨询

 400-617-6366
留言咨询
400-617-6366
留言咨询
 400-860-5168转2232
留言咨询
400-860-5168转2232
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP


