推荐厂家
暂无
暂无
同样的混标液10ug/mL(安赛蜜、苯甲酸、山梨酸、糖精钠、脱氢乙酸),同一根色谱柱C18柱,同样的色谱条件:(甲醇:0.02mol/L乙酸铵=5:95),流速1.0mL/min,在不同仪器下的分离度不同,效果还差挺多的:使用Waters2695e的糖精钠与脱氢乙酸分不开,而且脱氢乙酸还拖尾的厉害,而使用安捷伦1100的液相,这五个混标却分的不错,能实现基现分离,请问各位版友这个是什么原因造成的,
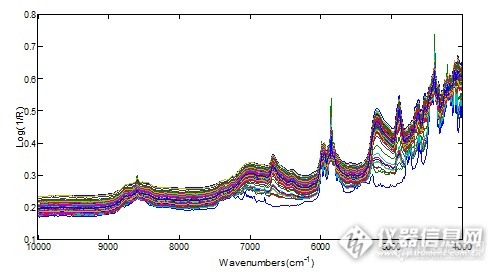
[align=center][b][url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术用于美洛西林钠舒巴坦钠药物混合过程在线混合均匀度终点监测[/b][/align][align=left][b]摘要: [/b]利用[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]技术,对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。[/align][b]关键词[/b]:[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url];分析模型;混合均匀度;在线监测自从2004年美国食品与药品监督管理局提出“过程分析技术”以来,全球的药品生产企业正在向着更高技术含量的生产方式和质量控制方式进军。近红外(Near infrared,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])光谱分析技术因其快速,无损的特点成为“过程分析技术”的重要组成部分,是制药企业进行产品中间体质量控制的重要方法之一。传统的检测方法为高效液相色谱法,紫外可见分光光度法等需要停止混合操作时才能取样检测,并且等待检测结果所需的时间也比较长,工作效率比较低,而[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱可以进行在线检测,连续记录不同混合时间内混合物的光谱图,建立数学模型对采集数据进行分析,从而判断各组分之间是否已经达到质量均一,工作效率大幅度的提高。本研究利用 [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] 光谱分析技术在线监测美洛西林钠舒巴坦钠的药物混合过程,从而实现混合终点的准确判断。[b]1 材料1.1试剂[/b]美洛西林钠(13102041,山东瑞阳制药有限公司)舒巴坦钠(SS201310-26,江西东风制药有限公司)[b]1.2仪器和软件[/b]AntarisII型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url](美国ThermoFisher公司),附有积分球采样模块;RESULT采样软件;电子分析天平(Sartorius BT224S,德国);TQ数据处理软件;表面皿;药匙;自制搅拌器。[b]2 方法2.1样品的准备[/b]精密称取舒巴坦钠固体原料药10.00g,美洛西林钠固体原料药40.00g,以备进行在线混合光谱的采集。平行制备3批样品,进行混合光谱的采集。[b]2.2模型的建立[/b]目前,[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于混合过程在线监测的方法可分为活性药物成分(API)定量分析模型监测和基于移动块标准偏差(MBSD)的定性分析模型监测。前者为基于API药物含量的定量监测模型,当达到混合终点时,API的含量趋于一定值,可以依据模型监测的含量是否达到理论值并趋于稳定进行混合终点的监测;后者为基于光谱的标准偏差的定性监测模型。MBSD法的基本原理为:连续采集的若干张光谱间的标准偏差变化率趋于稳定并小于限定的一阈值时可认为达到了混合终点。其具体的计算步骤为:首先确定用于计算光谱标准偏差的光谱的条数n(即移动块的宽度),当[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析仪器采集到n张光谱后计算n张光谱的峰面积(或最大峰高、平均峰高等)的标准差,当采集到n+1张光谱时将第一张光谱移除,计算最近n张光谱的标准差,如此类推,最终得到随时间变化的光谱的标准偏差,根据标准差的变化进行混合终点的监测。本研究中建立了舒巴坦钠含量的定量分析模型和基于MBSD法的定性分析模型同时对用于混合终点的判断。[b]2.3在线混合光谱的采集[/b]将称取的美洛西林钠、舒巴坦钠原料药样品放入表面皿中,然后将表面皿放在Antaris II型傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]积分球采样模块的上面,采用积分球漫反射采样方式进行光谱的采集。在运行在线混合工作流的同时采用自制的搅拌器进行样品的混合,采集得到混合过程的原始光谱,同时监测混合过程。波长范围10000-4000cm[sup]-1[/sup],每张光谱扫描次数4,混合过程中每间隔5s进行一张光谱的采集,光谱分辨率为8.0cm[sup]-1[/sup],每4个小时进行背景光谱的采集。每张[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱由1557个变量点组成。[b]2.4定量定性分析模型用于终点判断数据分析[/b]将在线混合过程进行监测,得到在线混合过程数据进行分析,以便了解混合全过程信息以及混合过程的监测。[b]2.5混合终点分析[/b]当得到混合终点时分别采集混合后的样品6处的原始[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,利用舒巴坦钠的定量分析模型预测混合终点时不同样品点处的舒巴坦钠的含量,判别是否混合均匀。[b]3 实验结果3.1分析模型的建立[/b]本研究中分别建立了在线混合过程的舒巴坦钠定量监测模型和基于移动块标准偏差的定性监测模型。[b]3.1.1 定性分析模型的建立[/b]目前混合均匀度在线监测常用的方法为MBSD法,本研究中MBSD法定性建模的参数为:选择的3个光谱区间包括全光谱、5275.6-4806.3cm[sup]-1[/sup](称为Region1)及7096.76-6344.66cm[sup]-1[/sup](称为Region2);用于计算光谱偏差的光谱的条数为5(即移动块的宽度为5)。[b]3.1.2 定量分析模型的建立[/b]本研究中所建立的定量分析模型用于监测混合过程中舒巴坦钠的百分含量的变化,因为本实验中舒巴坦钠和美洛西林钠两者间的混合比为4:1,当达到混合终点时,舒巴坦钠的百分含量应该在20%左右。其模型的具体参数见上一章中得到的舒巴坦钠百分含量的定量分析模型。[b]3.2混合在线过程数据分析[/b]本研究中平行进行了3次混合过程的在线监测,分别对3次实验结果进行分析,以充分了解混合监测过程。[b]3.2.1 第一批实验结果分析3.2.1.1 原始光谱图[/b]图1给出了混合过程中采集得到的208张原始光谱,由图中可知,处于下面的光谱较稀疏,可能属于混合刚开始的阶段,光谱会有较大的差异;处于上面的光谱较密集,其原因为随着混合的不断进行,光谱间差异越来越小,所以光谱较集中。[align=center][img=,498,274]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141912_01_1626619_3.png[/img][/align][align=center]图1 第一批混合过程原始光谱[/align][align=center] [/align][b]3.2.1.2 在线混合过程结果分析[/b]图2为定性分析模型中得到的3个光谱区间的峰面图,其中M1为全光谱建模的峰面积变化,M2为Region 1(5275.6-4806.3cm-1)的峰面积变化,M2为Region 2(7096.76-6344.66cm-1)的峰面积变化,由峰面积的变化图可知,混合过程的前100s其变化较为明显,M1不断升高,M2和M3(7096.76-6344.66cm-1)不断下降,之后峰面积值趋于稳定。[align=center][img=,525,234]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141913_01_1626619_3.png[/img][/align][align=center]图2 光谱区间峰面积图[/align]图3为舒巴坦钠含量及标准偏差变化图,由图中显示在混合的初期阶段,尤其是前100s左右,四个表征混合均匀度的参数均有着较大的变化趋势,在200-300s间四个参数有稍微较小的波动,此后随着混合过程的不断进行,表征混合均匀度的四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右,舒巴坦钠和美洛西林钠混合较为均匀,达到了混合终点。由图可知前100s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,538,292]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141914_01_1626619_3.png[/img][/align][align=center]图 3 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align][align=left] 当达到混合终点时分别采集表面皿下6个点的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,根据建立的模型测定其舒巴坦钠的百分含量,看混合是否均匀。表2给出了用所建模型得到的6个点的舒巴坦钠的百分含量值,6个点舒巴坦钠的百分含量值在20%左右,说明混合较为均一,但是最大的值达到了22.41%,可能是由于混合装置过于简陋,加上是人为搅拌进行混合,不能达到很好的混合,部分地方没有进行很好的混合。从实验的可行性方面,初步证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]技术用于美洛西林钠舒巴坦钠混合的可行性。[/align][align=center]表1混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,570,70]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_01_1626619_3.png[/img][/align][b]3.2.2 第二批实验结果分析3.2.2.1 原始光谱图[/b]图4给出了第二批混合过程中采集得到的203张原始光谱,其混合过程原始光谱的特征和第一批混合过程较为相似,混合初期光谱变化较为明显,随着混合的进行,光谱差异变小,光谱较为密集。[align=center][img=,488,280]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141915_02_1626619_3.png[/img][/align][align=center]图4 第二批混合过程原始光谱[/align][align=left] [b]3.2.2.2 在线混合过程结果分析[/b][/align]图5为各个光谱波段峰面积的变化图,由图中显示开始的100s内峰面积有着较大的变化幅度,随着混合的不断进行,峰面积的变化趋势不断减小并逐渐趋于稳定。[align=center][img=,516,307]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141916_01_1626619_3.png[/img][/align][align=center]图5 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图6为舒巴坦钠含量及标准偏差变化图,由图可知在混合的初期阶段大约0-100 s时,舒巴坦钠百分含量值及峰面积的标准偏差值有着明显的变化,全光谱峰面积的标准偏差(Full Range STD)在200-400 s间有较为明显的波段,此后随着混合过程的不断进行,四个参数变化范围均变小,模型给出的舒巴坦钠的百分含量在20%左右。由此可知前100 s是混合的主要阶段,此阶段舒巴坦钠的百分含量和标准偏差均有着明显的变化。[align=center][img=,551,327]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141917_01_1626619_3.png[/img][/align][align=center]图6 含量和标准偏差变化图[/align][align=center](a 舒巴坦钠百分含量 b 全光谱峰面积标准偏差 c Region 1峰面积标准偏差 d Region 2峰面积标准偏差)[/align]当达到混合终点时,采集表面皿底部6处的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱,检测混合过程是否达到均一,表2列出来了6处的舒巴坦钠的百分含量值,由表2可知达到混合结束后得到的6处的舒巴坦钠的百分含量均在20%左右,说明混合较为均匀。同时,由于实验条件的限制加上搅拌时人为因素的影响等,各点之间含量也着较大的差异。[align=center]表2 舒巴坦钠百分含量[/align][align=center] [img=,566,84]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141918_01_1626619_3.png[/img][/align][b]3.2.3 第三批实验结果分析3.2.3.1 原始光谱图[/b]图7给出了混合过程中采集得到的207张原始光谱,由图中可知,得到的原始光谱图与第一批和第二批有着相似的结果,即混合的初期光谱差异大,因此光谱较为稀疏(偏下方的光谱),随着混合的进行,光谱间差异变小,光谱变得密集(偏上方的光谱)。[align=center][img=,505,262]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_01_1626619_3.png[/img][/align][align=center]图7 第三批混合过程原始光谱[/align][b]3.2.3.2 在线混合过程结果分析[/b]图8给出了混合过程中3个光谱区间峰面积的变化趋势值,由图中可知0-100s间三个光谱区间的峰面积有着明显的变化,100-200s间峰面积有着明显的变化,但是变化幅度没有前100s大,200s以后峰面积变化趋势变小。说明前200s是混合的主要阶段,峰面积变化较为明显。[align=center][img=,519,343]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141919_02_1626619_3.png[/img][/align][align=center]图 8 光谱区间峰面积图[/align][align=center](a 全光谱峰面积 bRegion 1峰面积 cRegion 2峰面积)[/align]图9为舒巴坦钠百分含量及光谱峰面积的标准偏差随时间变化的趋势图,其变化趋势和峰面积的变化趋势相似,前100s变化幅度较大,100-200s间也有较为明显的变化,但是变化幅度不是很明显,200s后舒巴坦钠的百分含量和峰面积的标准偏差均趋于稳定,说明此时光谱差异变小,混合趋于均匀。[align=center][img=,529,352]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141920_01_1626619_3.png[/img][/align][align=center]图9 含量和标准偏差变化图[/align][align=center](a舒巴坦钠百分含量变化 b全光谱峰面积标准差 c Region1峰面积标准差 d Region2峰面积标准差)[/align]表3为达到混合终点时采集表面皿底部的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱得到的不同点的舒巴坦钠的百分含量值,由表中显示6个点的舒巴坦钠的百分含量值在20%左右,但是6个点之间舒巴坦钠百分含量间存在较大的差异,测得的最小值为17.80%,其原因可能是一方面由于实验条件的限制混合不够均匀,一方面用于舒巴坦钠含量测定的[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]定量分析模型也有一定的偏差,可能引起含量检测的差异存在。[align=center]表3 混合后不同点舒巴坦钠百分含量值[/align][align=center] [img=,564,66]http://ng1.17img.cn/bbsfiles/images/2017/09/201709141921_01_1626619_3.png[/img][/align][b]3.3小结[/b]通过3个混合平行实验的进行可知所建立的基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型能够有效的监测舒巴坦钠、美洛西林钠的混合过程。由舒巴坦钠百分含量和标准偏差变化图可知两者的变化有着相关性,当舒巴坦钠的百分含量变化幅度大时,其标准偏差的变化幅度也较大,因此两者均可以用于混合过程的在线监测,证实了实验的可行性。[b]4 结论和讨论[/b]本研究采用AntarisII傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]对美洛西林钠、舒巴坦钠混合过程进行了在线监测。在研究中,分别建立了基于MBSD法的定性分析模型和基于舒巴坦钠百分含量的定量分析模型,然后Antaris II傅里叶变换[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱仪[/color][/url]漫反射采样方式采集混合过程中的光谱,实时监测混合过程的进行。通过3个平行实验的在线混合过程,结果显示MBSD法和舒巴坦钠百分含量测定法均能有效的监测其混合过程,有效的证明了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术用于舒巴坦钠、美洛西林钠混合在线监测的可行性。此外,MBSD法因为无需进行一级数据的采集,方法较为简单且容易理解,目前常用于混合过程的在线监测。本研究中有效证实了[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]光谱分析技术在舒巴坦钠美洛西林钠样品在线混合过程中应用的可行性,在样品的在线混合监测中有着重要的应用价值和应用前景。该技术能够克服传统方法费时、繁琐等缺点,而且可以实现过程的实时在线监测,让生产者充分了解整个生产过程中的参数变化。 [b]参考文献[/b]陆婉珍, 褚小立. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]([url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url])和过程分析技术(PAT). 现代科学仪器, 2007(004):13-17.SieslerH, Ozaki Y, Kawata S, et al. Near-infrared spectroscopy: principles .Instruments, Applications, 2002:35-181.Bhushan,K.R.,et al.Detection of breastcancer microcalcifications using a dual-modality SPECT/[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url] fluorescent probe. J Am Chem Soc, 2008. 130(52):17648-17649.贾燕花. [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术在化学药品生产过程控制应用初探. 北京协和医学院, 2011.Fevotte.G,et al.Applications of [url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]NIR[/color][/url]spectroscopy to monitoring and analyzing the solid state during industrialcrystallization processes . Int J Pharm, 2004, 273(1):159-169.张敏.盐酸林可霉素多晶型分子构象对其红外光谱行为的影响.中国抗生素杂志, 2005, 30(009):529-532.Blanco M,R Goz"01ez Ba,E.Bertran,Monitoring powder blending in pharmaceutical processes by use of nearinfrared spectroscopy . Talanta, 2002, 56(1):203-212,田科雄.不同装载系数和混合时间对添加剂预混料混合均匀度的影响.河北畜牧兽医, 2004, 20(9):52-53.孙栋. 基于[url=https://insevent.instrument.com.cn/t/1p][color=#3333ff]近红外光谱[/color][/url]分析技术的几种固体粉末混合均匀度快速检测研究. 山东大学硕士学位论文, 2012年.
(1)细胞核的分离: 将培养的细胞制成细胞混悬液,或以胰蛋白酶消化法于培养盖上收集培养细胞。应用MgSO4染色分离方法分离细胞核(Van den Engh et al 1986, 在Traok和Van Den Engh 34章 )。细胞混悬液浓度为5×106/ml。利用RNA酶消化后,使核从细胞分离,细胞核混悬液浓度为4~5×106细胞核/ml。(2)细胞固定和酸的处理:在5 ml试管内加冷的100%酒精不断旋转以达到满意的固定。在冰上停留10 min。在4 ℃离心(×150 g)10 min。重复加三次冷的100%酒精入试管内,离心,倾去。置于冰上10 min,再离心。然后加入相当核悬液1/2量的0.1 n HCl , 0.5% Triton X-100。室温停留10 min。加入IBM-0.25%Triton X-100(IBM配方:50 mmol/l KCl, 10 mmol/L MgSO4, 5 mmol/L HEPES pH8.0)。再离心,重复IBM漂洗(这时细胞核可在不染色情况下,以荧光显微镜观察)后,以2×SSC-0.1%Tween漂洗1×3min,继之加入等量2%的多聚甲醛在1×PBS-5 mmol/l MgSO4。在室温静置站立10min。倾去上清液,加IBm -Triton X-100漂洗,离心,使细胞核混悬液最终浓度为108/ml,(可用IBM-Triton X-100稀释约50倍,在血球计数器计数),混悬液镜检应含单个,完整的细胞核。(3)细胞核混悬液杂交①配制杂交混合液:甲酰胺5份,20×SSC1份,50%硫酸葡聚糖2份,pH调至7.0。此原液(stock solution)可贮存在4 ℃冰箱内。应用时加1份10 mg/ml鲱鱼精子DNA(herring sperm DNA)。②混合1 μl的细胞核混悬液(108/ml)与18μl的杂交混合液,充分混匀。将此19 μl混合液移入1.5 ml容积的Eppendorf 管中(核含量约为105)。③加入100 ng/每管的AAF标记DNA探针(如为生物素标记DNA探针浓度为20~40 ng/每管)。④置70 ℃10min使DNA探针和核DNA变性。⑤和组织切片与DNA探针杂交方法相较,不同的是在加热变性后切勿置冰上迅速冷却以终止反应,而应迅速转入37 ℃孵育过夜。(4)杂交后漂洗在每管中加入1.25 ml 50%甲酰胺-2×SSC(pH7.0),在42 ℃静置10~15min。偶尔旋转以助混匀。冷却至室温。加100 μl经dimethylsuberimidate(DEMS)处理的血细胞(107/ml)混匀,离心,室温,10min,轻弹试管使沉淀的小块散开,加入1.25 ml 2×SSC(pH7.0),42 ℃,继之,静置于室温10~15min,如前离心,再加1.25 ml IBM-Triton X-100,室温静置5min,离心。注:DEMS处理红细胞方法:经漂洗并离心去除白细胞和血清的红细胞在盐液如PBS中,细胞含量为108/ml,以K2CO3和DEMS溶液处理3次,第1次:K2CO3为20 mmol/L,DEMS为3 mmol/L,以后2次:K2CO3依然为20 mmol/L,而DEMS为10 mmol/L。在应用前将K2CO3和DEMS液混合加入红细胞混悬液中。在最后2次漂洗液中,应用100 mmol/l K2CO3将pH调至9~10。在25 ℃,15min后,加入50 μl,100 mmol/l 的柠檬酸(citric acid)/每ml细胞混悬液的浓度以达固定红细胞的目的。固定的红细胞离心倾去上清液后,用2×SSC稀释到108/ml,加0.1%叠氮钠可在4 ℃保存至少1年。(5)AFF标记的荧光显示:加200 μl的PBS含0.05%Tween和2%正常血清(NGS),轻轻振荡混匀,室温静置10min,加20 μl 1:100的单克隆抗AFF抗体,37 ℃孵育45min,加1.25 ml的PBS-Tween,室温静置10min,加20间歇性振荡,离心,倾去上清液,加200 μlPBS含0.05%Tween –2%NGS,振荡,室温静置10min,加20 μl的羊抗小鼠–FITC荧光标记抗血清,稀释度1:100~1:300。孵育于37 ℃ 45min,加1.25 ml PBS –Tween,室温静置10min,离心,倾去上清液。(6)生物素标记探针的荧光显示:加200 μl 4×SSC含0.1%Trion X-100 和5%BSA。室温静置10 min后,加20 μl抗生物素标记FITC抗血清15 μg/ml,孵育在37 ℃ 30min,以1.5 ml 4×SSc –0.1% Trion X-100洗1次,加入1.25 ml IBm –Triton X-100,室温静置10~15min,间歇振荡、离心。(7)荧光显微镜观察:将细胞核混悬液稀释于250 μl的IBM-Trion X-100中,轻加振荡混匀。为抗荧光褪色可加等量的抗褪色溶液至载片上的细胞核涂片上,选择适当的激发波长观察。(8)流式细胞计:将750 μl的细胞核混悬液通过流式细胞仪(Flow cytometry, FCM),DEMS处理过的红细胞作为对照(Df 530/30nm, Omega ·Optical Inc, Brattleboro, VT)。





 400-611-9236
400-611-9236
 留言咨询
留言咨询
 400-875-8210
留言咨询
400-875-8210
留言咨询
 400-875-8210
留言咨询
400-875-8210
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP


