推荐厂家
暂无
暂无
最近要开展测烤鳗中甲苯咪唑残留的检测,请问有国标法或是SN法吗?我的Email:capinter@163.com,谁手头有能发一份给我吗?,先谢过了~~~
求芝麻酚,咪唑和二甲苯麝香检测的标准。国标,国外的都可以,主要是标准!谢谢!
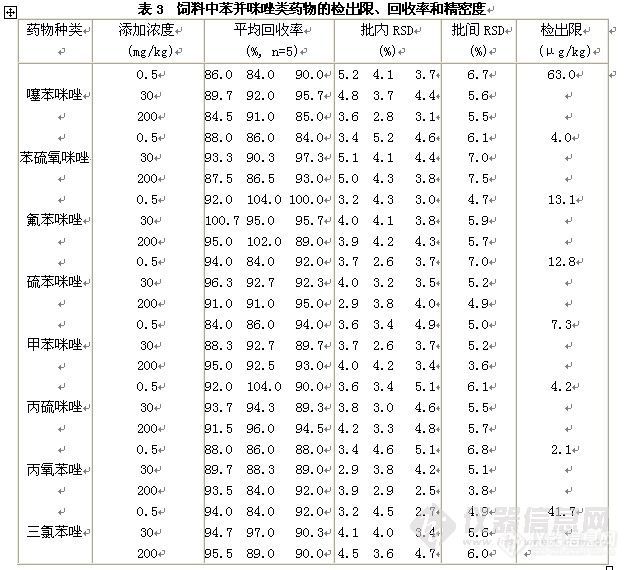
液相色谱串联质谱法测定饲料中8种苯并咪唑类药物摘 要 建立了同时测定饲料中8种苯并咪唑类药物(噻苯咪唑、丙硫咪唑、硫苯咪唑、苯硫氧咪唑、氟苯咪唑、甲苯咪唑、丙氧苯唑和三氯苯唑)的液相色谱串联质谱分析方法。饲料样品直接用酸化乙腈提取,提取液用甲酸溶液稀释后直接进行分析。分析时采用XBridgeTM C18色谱柱,以甲酸溶液-乙腈体系进行梯度洗脱,MRM方式测定,基质外标法定量。苯并咪唑类药物在0.02~10 mg L-1浓度范围内呈良好的线性,线性相关系数均大于0.990,苯并咪唑类药物在饲料样品中最低检测限为2.1~63.0μg/kg。饲料中苯并咪唑类药物在0.50~200 mg/L范围内的回收率为84.0%~104%之间,相对标准偏差(RSD)均小于10%。 关键词 苯并咪唑类药物;液相色谱串联质谱法;饲料 苯并咪唑类药物(benzimidazoles, BMZs)属于广谱、高效、低毒抗蠕虫药,由于对胃肠线虫具有很强的驱杀作用,至今仍在广泛使用。但由于BMZs在实验动物和靶动物显示致畸和致突变作用,目前使用的BMZs多数是食品残留中重要的监控对象,且BMZs在体内转化的代谢产物仍具有毒理作用,所以我国以及联合国粮农组织、欧盟、美国、日本等国家和组织都将苯并咪唑类药物列入限制使用的兽药药物,并制订出各种苯并咪唑类药物在不同动物体内(肌肉、组织、奶等)的最高残留限量。饲料安全直接关系到动物性食品的安全,考虑到苯并咪唑类药物经常被添加到饲料中使用,故很有必要进行饲料中苯并咪唑类药物的分析研究。 目前对于动物组织中苯并咪唑类药物的分析方法较多,而饲料中苯并咪唑类药物分析方法国内未见发表,国外也较少,涉及的种类也较少,最多的仅有5种药物。动物组织和饲料中BMZs分析涉及的主要分析手段有:酶联免疫吸附法( ELISA) 、气相色谱-质谱法(GC-MS)、高效液相色谱法(HPLC)及高效液相色谱串联质谱法(HPLC-MS/MS),高效毛细管电泳法(HPCE)。考虑到苯并咪唑类药物在我国使用情况,本研究选择了8种常用苯并咪唑类药物,考虑到LC-MS/MS法灵敏度高的特点,样品酸化乙腈提取后直接稀释后进行液相色谱串联质谱分析。1 材料与方法1.1 仪器与试剂 Waters 2695 Quattro MicroTM API高效液相色谱串联质谱仪(美国Waters公司),配置电喷雾离子源;固相萃取仪(美国Supelco 公司);Sigma离心机。噻苯咪唑和丙硫咪唑标准品(Accustandard 公司);硫苯咪唑、苯硫氧咪唑、氟苯咪唑、甲苯咪唑、丙氧苯唑和三氯苯唑标准品(Dr. Ehrenstorfer)。乙腈、二甲亚砜和甲酸为色谱纯(Fisher公司)。1.2 仪器条件 XBridgeTM C18色谱柱(150 mm×2.1 mm,内径3.5 μm);流动相A为0.1%甲酸溶液,B相为乙腈,梯度洗脱条件:B相在1.0 min内从15%线性增加到25%,再在2.5 min内线性增加到95%,保持3.5 min,然后在0.1 min内降至15%,保持4.9 min;流速:0.3 mL/min;进样量:10 µL;柱温:30℃。 质谱条件:ESI源正离子模式电离;多反应监测(MRM);毛细管电压:3.0 KV;萃取锥孔电压:20 V;RF透镜电压:0.5 V;离子源温度:110 ℃;脱溶剂气温度:350 ℃;锥孔气流速:50 L/h;脱溶剂气流速:600 L/h;倍增器电压:650 V;二级碰撞气:氩气;其它条件详见表1。http://ng1.17img.cn/bbsfiles/images/2010/11/201011301506_262957_1759541_3.jpg1.3 样品处理 称取2g试样(精确到0.01g)于50 mL离心管中,加入20 mL0.5 %甲酸乙腈,涡旋1 min,然后超声提取10 min,以5000 r/min的速度离心5 min后吸取1.0 mL上清液于5 mL刻度试管中,加入3 mL0.1 %甲酸溶液于试管中,混匀后过0.22 μm滤膜,进行液相色谱串联质谱分析。1.4 线性实验 准确称取各10.0 mg BMZs标准品于相应的10mL容量瓶中,噻苯咪唑、甲苯咪唑、丙氧苯唑和丙硫咪唑用二甲亚砜溶解并定容至刻度,其余4种BMZs用甲醇:二甲亚砜(2:3 v/v)溶解并定容至刻度,即得均为1000 mg/L标准储备液。分别吸取1.0 mL各标准储备液于同一10mL容量瓶中,用甲醇稀释至刻度,即得100 mg/L的混合标准工作液。分别准确移取苯并咪唑类药物混合标准中间液适量,配制浓度为0.2.、0.8、2.0、10.0、40.0和100.0 mg/L的系列标准溶液,吸取0.1 mL于5 mL刻度试管中,再吸取空白试料提取液0.9 mL于该5 mL刻度试管中,加入3 mL0.1%甲酸溶液后混匀过膜,进行上机测定,以定量离子对峰面积为纵坐标,标准溶液浓度为横坐标,绘制基质校准标准曲线。2 结果与分析2.1 液相色谱质谱分析 苯并咪唑类药物色谱分析时,通常采用反相分离体系,主要有三类流动相体系:离子增强体系,pH2~3,一般使用乙腈-磷酸或磷酸盐体系;离子抑制流动相体系,pH5~7;离子对流动相体系,离子增强流动相中加入阴离子对试剂。对于多组分苯并咪唑类药物液相色谱质谱分析时,通常采用离子增强体系进行梯度洗脱,如0.1%甲酸溶液-乙腈体系,因为该体系和纯水-乙腈体系相比色谱峰的拖尾现象得到了明显改善。 苯并咪唑类药物属弱碱性物质,中等极性,在酸性条件下很容易质子化,于是本方法选择ESI+进行分析。以乙腈/0.1%甲酸溶液(3:7,v/v)为溶解液,用蠕动泵(20μL/min)对苯并咪唑类药物的质谱条件进行优化。经过优化的条件为:毛细管电压:3.0KV;离子源温度:110℃;脱溶剂气温度:350℃;锥孔气流速:50L/h;脱溶剂气流速:600L/h。其它条件详见表1。2.2 提取净化方法的选择和优化 [font=宋体






 留言咨询
留言咨询
 留言咨询
留言咨询
 留言咨询
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP


