推荐厂家
暂无
暂无
(按笔划顺序排列) 八角莲、八里麻、千金子、土青木香、山莨菪、川乌、广防己、马桑叶、马钱子、六角莲、天仙子、巴豆、水银、长春花、甘遂、生天南星、生半夏、生白附子、生狼毒、白降丹、石蒜、关木通、农吉痢、夹竹桃、朱砂、米壳(罂粟壳)、红升丹、红豆杉、红茴香、红粉、羊角拗、羊踯躅、丽江山慈姑、京大戟、昆明山海棠、河豚、闹羊花、青娘虫、鱼藤、洋地黄、洋金花、牵牛子、砒石(白砒、红砒、砒霜)、草乌、香加皮(杠柳皮)、骆驼蓬、鬼臼、莽草、铁棒槌、铃兰、雪上一枝蒿、黄花夹竹桃、斑蝥、硫磺、雄黄、雷公藤、颠茄、藜芦、蟾酥。
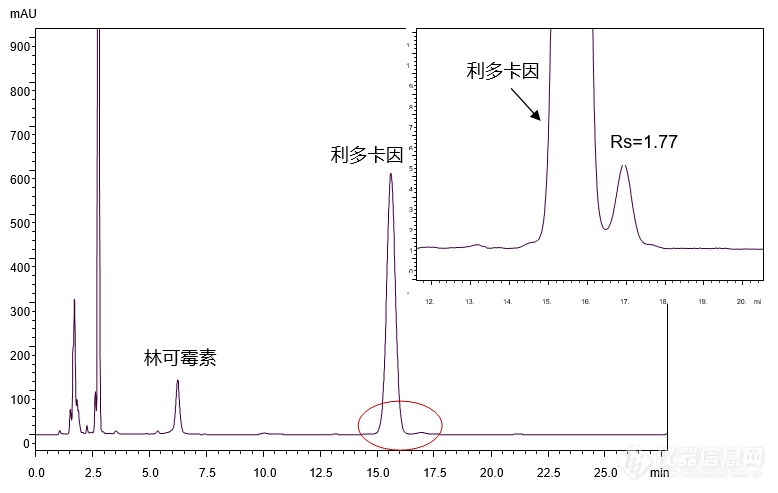
[align=center][b]【国家药品标准】林可霉素利多卡因凝胶的分析[/b][/align][align=center][b][/b][/align][align=right][b]——依据国家药品标准WS-10001-(HD-0140)-2002方法[/b][/align][b]林可霉素利多卡因凝胶[/b]为复方制剂,每克含林可霉素5毫克,利多卡因4毫克。适应症为用于轻度烧伤、创伤及蚊虫叮咬引起的各种皮肤感染。 [img=,193,127]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834522166_2994_2222981_3.gif!w193x127.jpg[/img] [img=,140,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834520028_3541_2222981_3.gif!w140x64.jpg[/img] 林可霉素 利多卡因 Lincomycin Lidocaine M.W.: 406.54 M.W.: 234.34客户提供林可霉素利多卡因凝胶样品,希望本实验室帮忙通过筛选色谱柱及调节分析条件,依据[color=#ff0000][b]国家药品标准WS-10001-(HD-0140)-2002[/b][/color]方法,实现林可霉素利多卡因凝胶样品的良好分析。首先,使用能在纯水条件下稳定使用的高极性色谱柱[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 150 mm[/b][/color],对林可霉素利多卡因凝胶样品进行分析,结果如图1所示,[color=#330099]利多卡因与其峰后杂质之间分离度为1.77[/color]。[align=center][img=,690,437]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858200006_8607_2222981_3.png!w690x437.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]AQ分析所得色谱图[/align]注:峰上标数字为分离度。[img=,528,205]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858202566_2695_2222981_3.png!w528x205.jpg[/img]为进一步提高利多卡因与其峰后杂质之间的分离度,在原条件基础上将柱温由30℃降低至25℃,并分别使用 CAPCELL PAK C[sub]18[/sub] AQ、CAPCELL PAK C[sub]18[/sub] MG及高含碳量ODS色谱柱SUPERIOREX ODS进行分析,结果如图2所示。[align=center][img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859201516_7229_2222981_3.png!w690x490.jpg[/img][/align][align=center]图2 25℃条件下不同色谱柱分析结果对比[/align]注:峰上标数字为分离度。[img=,637,223]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859204236_7198_2222981_3.png!w637x223.jpg[/img]如图2所示,在柱温25℃条件下使用三款色谱柱进行分析,其中,[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ色谱柱分析结果最好,利多卡因与其峰后杂质分离得到最佳分离,分离度为4.23[/b][/color];[color=#330099][b]使用CAPCELL PAK C[sub]18[/sub] MG色谱柱进行分析时,利多卡因与其峰后杂质分离度为3.27[/b][/color];而使用SUPERIOREX ODS色谱柱分析时,利多卡因与其峰后杂质未得到有效分离。综上,在国家药品标准WS-10001-(HD-0140)-2002方法基础上,将色谱柱柱温由30℃降低至25℃,使用高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ及中等极性色谱柱CAPCELL PAK C[sub]18[/sub] MG进行分析,均可在25 min内完成林可霉素利多卡因凝胶样品的分析,并得到利多卡因与其峰后杂质之间的良好分离结果。[align=right][/align][align=right][/align][align=right] [/align][align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
标准曲线法是仪器分析中最常用的方法,从标准来看GB/T22554-2010《基于标准样品的线性校准》中:1、标准曲线的浓度范围应覆盖正常操作条件下的被测量范围;2、标准样品的组分尽量与被测样品组分一致;3、标准样品的浓度值应等距离的分布在被测量范围;4、标准样品的个数至少应有3个浓度;5、每个标准点至少重复2次,这个重复是指从稀释开始;如果国家标准有相应的浓度系列推荐,尽量按国家标准。工作中我们经常采用线性校准,因为线性方程最为简洁。 标准曲线需要几个数据点,是由所检测组分的浓度范围、分析仪响应特性、干扰因素、浓度与检测信号响应类型有关的。 3个点:对于一些低浓度,特别是微量分析,并且浓度范围不是很大的,检测器响应可靠,背景干扰非常小的,则可以选用较少的工作点就行,一般有3个浓度点就足够了,有些甚至可以只用一个浓度点就行,另一个点直接用坐标原点。 4~6 个点:对于平时测量的样品浓度范围较宽,并且检测器响应不完全是一次曲线(直线)的,可以采用二次曲线或分段校正方式,以减少数据偏差,这种情况就需要多用几个数据点,如果不分段,数据点有4~6 个就够用。但如果是分段校正,则每段需要至少两个数据点。而对于一些样品无浓度范围规律的,特别是一些检测机构,如果分析仪的检测响应可靠,环境因素影响少的,可以做校正曲线。若是环境因素影响大的,则不必做校正曲线,而采用标准加入法或标准加入-一次稀释法反而简便一些。 实际上,一般是做五个点(不包括零浓度);一般以检出限的5~10倍为第一个点,以后根据1倍(或接近一倍)递增,最高浓度是最低浓度的10~20倍为宜。当然,要根据仪器的灵敏度来调整。 标曲R值是几个9才合格? 实验室应按检测标准(方法)的要求使用标准溶液或标准物质建立标准曲线。所用标准溶液或标准物质应覆盖被测样品的浓度范围。检测标准(方法)如无具体的要求,至少使用5个标样(除空白外)建立线性标准曲线,每个点重复测定1-3次,对于筛选方法,线性回归方程的相关系数不应低于0.98,对于确证方法,相关系数不应低于0.99。其实〉0.99是判断是否为线性相关的一个标准,实际应用中线性 〉0.999才是比较理想的。线性在0.99到0.999之间的监测结果只用接近最高浓度一半(中间浓度)的位置才比较准确,如果线性大于0.999的话,在整个线性范围内都会有一个比较满意的结果。如果检测的线性不好,可以减少标准的覆盖范围,将标准的浓度调整到待测样品浓度附近,这样结果也是非常准确的。例如,样品的浓度约20ppb,但在0~50ppb范围建立标准曲线,但线性非常不理想,这时可以将标准范围调整到15~25ppb之间,作五个标准。标准曲线建立后应在样品的检测中消除空白造成的影响。高于接受限的试剂空白表示与空白同时分析的这批样品可能受到污染,检测结果不能被接受。标准曲线建立后,必要时应在分析样品前加标,添加物浓度水平应接近分析物浓度或在校准曲线中间范围浓度内,加入的添加物总量不应显著改变样品基体。标准曲线有效期到底规定:(1)标准曲线属于实验室质量控制的范围,按照《实验室资质认定评审准则》中结果控制的要求:定期使用有证标准物质(参考物质)进行监控和/或使用次级标准物质(参考物质)开展内部质量控制。(2)准则中并未对“定期”进行规定,所以如果“定期”,就根据实验室的实际情况来定了。(3)既然准则上没说明,那根据一般的分析教材,当实验条件(包括药剂、人员、仪器等)发生变化时,最好重新制作标准曲线。一般来说仪器如果长期使用,并经过检定,是处于稳定状态,而人员药剂的变化往往会较大。如果说每次换个人操作都要换曲线,那工作量就太大了。每次开机都必须做标准曲线?从药品检测的要求来说,因为:1、每天所配的流动相都会有所不同,导致出峰的时间都会有一定的差异,峰面积相应都有所差异。2、检测器光能也在不断的衰弱,因此其每天的相应值也有所不同,其峰面积也有差异。基于以上原因,原则上应该是每天都要进行标准曲线校正的。标准曲线不要每次都做,但是每次必须进标准品样品;因为每次你的流动相和原来的不可能完全一样,同时仪器的状态也在变化,所以不同批次间的保留时间是不一致的,所以你必须用随行标准品来定位与定量;标准曲线不一定非要过原点,只要线性好就可以了。因为做空白的话基本上不会过原点。过原点是强制过的,实际曲线可能不过,就会造成误差。不过原点是因为有系统误差。不过系统误差的话,大家都误差也就抵消了,没有太大影响,针对是否过原点(0,0)问题,可以从原点(0,0)代表的意义来考虑:即所测组分浓度为零的时候,信号响应值(液相色谱也就是峰高或者峰面积)是否也为零。通常来说色谱分析是属于这种情况的,因而可以把(0,0)点作为一个浓度水平计入标准曲线(甚至不需要真的去配一个浓度为零的标准溶液来进样),这也是单点法定量的一个依据;标准曲线的线性范围线性范围,主要从相关系数r看,一般要求r大于等于三个九。之前做实验有时候浓度高时,线性不好,高浓度点不在标准曲线上,而是在标准曲线的下面,而且离拟合的标准曲线比较远。遇到这种情况,标准曲线的线性相关系数就很差,有时候才一个九,,如果自己用手动拟合的话,用平滑的曲线去连接所有点的话,你就会发现,如果在线性范围内,连接起来就是直线,如果超出了线性范围,连接起来就是一条弯曲的曲线。标准曲线的相关系数的有效数字该如何保留呢?GB5750.3-2006 8.2.7项有如下规定:校准曲线相关系数只舍不入,保留到小数点后出现非9的一位,如0.99989→0.9998。如果小数点后都是9时,最多保留小数点后4位。小结标准曲线法有一定的优势,也有一定的缺陷,它特别适合于大量样品的分析。但由于每次样品分析的色谱条件(检测器的响应性能,柱温,流动相流速及组成,进样量,柱效等)很难完全相同,因此容易出现较大误差。此外,标准工作曲线绘制时,一般使用欲测组分的标准样品(或已知准确含量的样品),而实际样品的组成却千差万别,因此必将给测量带来一定的误差。






 400-858-8867
400-858-8867
 留言咨询
留言咨询
 400-858-8867
留言咨询
400-858-8867
留言咨询
 400-860-5168转4124
留言咨询
400-860-5168转4124
留言咨询









 我要推广仪器
我要推广仪器
 下载APP
下载APP


