

方案详情文
智能文字提取功能测试中
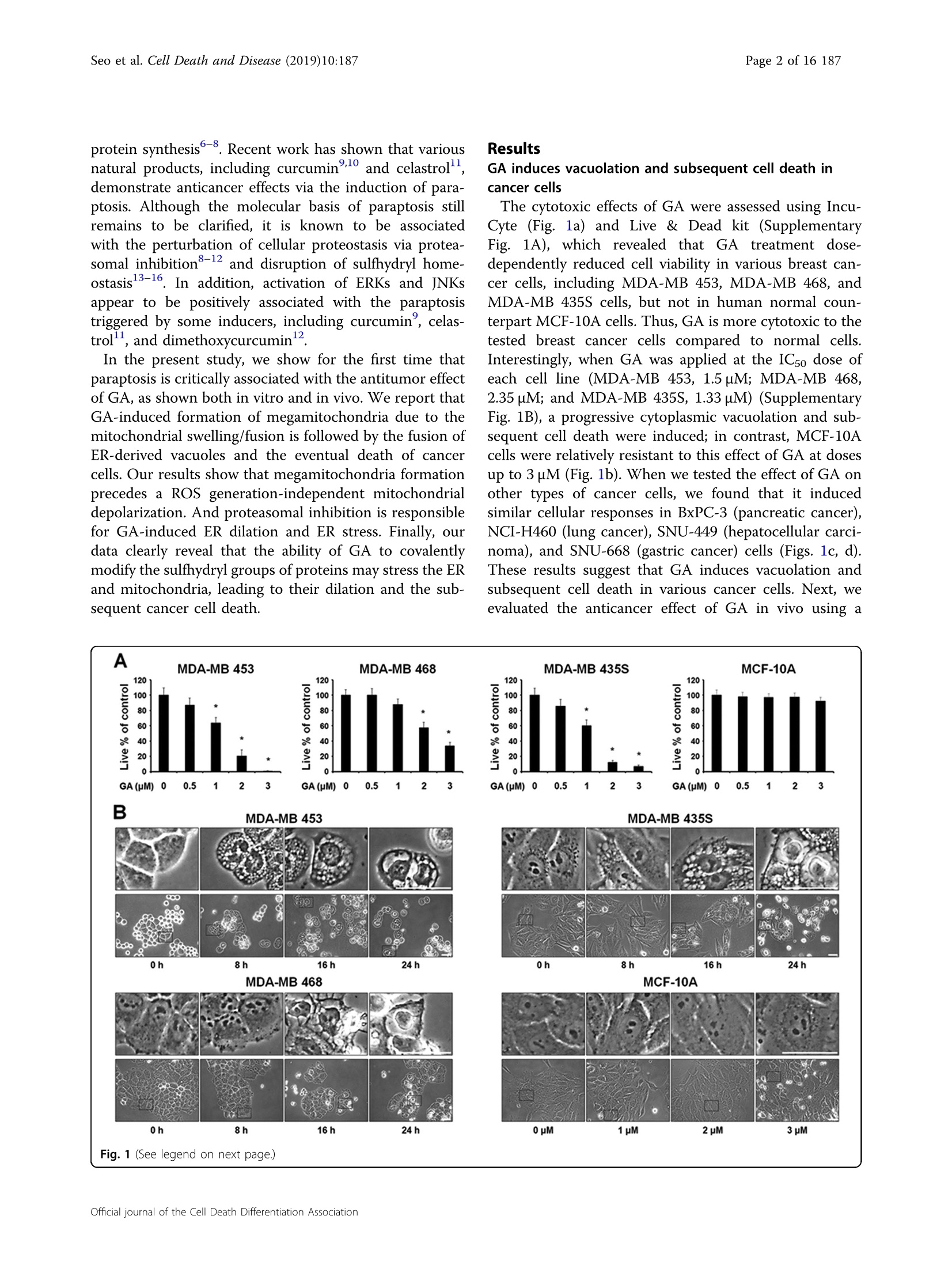
Seo et al. Cell Death and Disease (2019)10:187https://doi.org/10.1038/s41419-019-1360-4Cell Death & DiseaseARTICLE Seo et al. Cell Death and Disease (2019)10:187Page 2 of 16 187 Open Access Gambogic acid triggers vacuolization-associated cell death in cancer cells viadisruption of thiol proteostasis Min Ji Seol, Dong Min Leel, In Young Kim, Dongjoo Lee, Min-Koo Choi, Joo-Youn Lee, Seok Soon Park,1,2Seong-Yun Jeong, Eun Kyung Choi and Kyeong Sook Choil2 Abstract Gambogic acid (GA), a xanthonoid extracted from the resin of the tree, Garcinia hanburyi, was recently shown to exertanticancer activity in multiple studies, but the underlying action mechanism remains unclear. Here, we show that GAinduces cancer cell death accompanied by vacuolation in vitro and in vivo. This GA-induced vacuolation in variouscancer cells was derived from dilation of the endoplasmic reticulum (ER) and mitochondria, and was blocked bycycloheximide. These findings suggest that GA kills cancer cells by inducing paraptosis,a vacuolization-associated celldeath. We found that megamitochondria formation, which arose from the fusion of swollen mitochondria, precededthe fusion of ER-derived vacuoles. GA-induced proteasomal inhibition was found to contribute to the ER dilation andER stress seen in treated cancer cells, and megamitochondria formation was followed by mitochondrial membranedepolarization. Interestingly, GA-induced paraptosis was effectively blocked by various thiol-containing antioxidants,and this effect was independent of ROS generation. We observed that GA can react with cysteinyl thiol to formMichael adducts, suggesting that the ability of GA to covalently modify the nucleophilic cysteinyl groups of proteinsmay cause protein misfolding and subsequent accumulation of misfolded proteins within the ER and mitochondria.Collectively, our findings show that disruption of thiol proteostasis and subsequent paraptosis may critically contributeto the anti-cancer effects of GA. Introduction The identification and development of effective antic-ancer agents with fewer side effects is critical for thesuccessful management of cancer. Gambogic acid (GA) isa xanthone structure isolated from the dry, brownishgamboge resin of Garcinia hanburyi. GA has been shownto confer potent anticancer activity via multiple effects indifferent types of cancers, including inhibition of cancercell proliferation, induction of apoptosis, inhibition ofangiogenesis and inhibition of metastasis . Importantly, Correspondence: Kyeong Sook Choi (kschoi@ajou.ac.kr)Department of Biomedical Sciences, Ajou University Graduate School ofMedicine, Suwon 16499, KoreaDepartment of Biochemistry and Molecular Biology, Ajou University, Suwon 16499, Korea Full list of author information is available at the end of the article.Edited by C. Munoz-Pinedo GA exhibits more toxicity toward cancer cells than tonormal cells134. A phase IIb trial of GA is currently beingundertaken to test efficacy of GA in treating non-smallcell lung, renal and colon cancers. Malignant cancer cells often show innate and acquiredresistance to apoptosis; thus, alternative means to combatmalignant cancer via non-apoptotic cell death may offer anattractive therapeutic strategy to effectively kill malignantcancer cells that resist conventional pro-apoptotic cancertherapies. In this study, we show for the first time that GAcan kill various cancer cells via the induction of paraptosis.Paraptosis is a programmed cell death mode that ischaracterized by dilation of the ER and/or mitochondria°.It lacks apoptotic features, including chromatin con-densation, DNA fragmentation, apoptotic body formationand caspase dependency, and is known to require de novo ◎ The Author(s) 2019 O.cpen Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing,adaptation, distribution and reproduction@ in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate ifchanges were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. Ifmaterial is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtainpermission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/. protein synthesis8 Recent work has shown that variousnatural products, including curcumin’10 and celastrol,demonstrate anticancer effects via the induction of para-ptosis. Although the molecular basis of paraptosis stillremains to be clarified, it is known to be associatedwith the perturbation of cellular proteostasis via protea-somal inhibition8-12 and disruption of sulfhydryl home-ostasis 3-16. In addition, activation of ERKs Ksappear to be positively associated with the paraptosistriggered by some inducers, including curcumin’, celas-trol, and dimethoxycurcumin. In the present study, we show for the first time thatparaptosis is critically associated with the antitumor effectof GA, as shown both in vitro and in vivo. We report thatGA-induced formation of megamitochondria due to themitochondrial swelling/fusion is followed by the fusion ofER-derived vacuoles and the eventual death of cancercells. Our results show that megamitochondria formationprecedes a ROS generation-independent mitochondrialdepolarization. And proteasomal inhibition is responsiblefor GA-induced ER dilation and ER stress. Finally, ourdata clearly reveal that the ability of GA to covalentlymodify the sulfhydryl groups of proteins may stress the ERand mitochondria, leading to their dilation and the sub-sequent cancer cell death. Results GA induces vacuolation and subsequent cell death incancer cells The cytotoxic effects of GA were assessed using Incu-Cyte (Fig. 1a) and Live & Dead kit (SupplementaryFig. 1A), which revealed that GA treatment dose-dependently reduced cell viability in various breast can-cer cells, including MDA-MB 453,MDA-MB 468, andMDA-MB 435S cells, but not in human normal coun-terpart MCF-10A cells. Thus, GA is more cytotoxic to thetested breast cancer cells compared to normal cells.Interestingly, when GA was applied at the ICso dose ofeach cell line (MDA-MB 453, 1.5 uM; MDA-MB 468,2.35 uM; and MDA-MB 435S, 1.33 pM) (SupplementaryFig. 1B), a progressive cytoplasmic vacuolation and sub-sequent cell death were induced; in contrast, MCF-10Acells were relatively resistant to this effect of GA at dosesup to 3 uM (Fig.1b). When we tested the effect of GA onother types of cancer cells, we found that it inducedsimilar cellular responses in BxPC-3 (pancreatic cancer),NCI-H460 (lung cancer), SNU-449 (hepatocellular carci-noma), and SNU-668 (gastric cancer) cells (Figs. 1c, d).These results suggest that GA induces vacuolation andsubsequent cell death in various cancer cells. Next, weevaluated the anticancer effect of GA in vivo using a Fig. 1 GA induces cell death accompanied by vacuolation in vitro and in vivo. a, c Cells were treated with the indicated concentrations of GAfor 24 h. Cellular viability was assessed using IncuCyte as described in Materials and Methods. Data represent the means±SEM (n=3). Statisticalsignificance was determined usingone-way ANOVA followed by Bonferroni's post hoc tests. *p<0.01 vs. untreated control. b, d Cells treated with GAat the concentrations around the IC50,(MDA-MB 453 (1.5 uM), MDA-MB 468 (2.35 uM), MDA-MB 435S (1.33uM), BxPC-3 (1.68uM), NCI-H460 (2.35uM),SNU-449 (1.62 uM), and SNU-668 (1.41 pM)), which were calculated using GraphPad Prism, were observed by phase-contrast microscopy. MCF-10Acells treated with GA at the indicated concentrations for 24 h were observed by phase-contrast microscopy. Bars, 40 um. e Athymic nude mice of6-8 weeks old were xenografted with MDA-MB 435S cells and injected with vehicle, 4 mg/kg GA, and 8mg/kg GA as described in Materials andMethods.Tumor sizes were measured every 2-3 days after the beginning of vehicle or GA injection and plotted for growth curve. Data represent themeans± SD. Kruskal-Wallis test was performed followed by Dunn’s test. *p <0.05 vs. vehicle-treated mice. f treated mice and tumors isolated fromthose mice were photographed on the 14th day. g The results of H&E staining in tumor tissues of the mice treated with 4 mg/kg GA. Bars, 20 um MDA-MB 435S cell xenograft model. Mice receivedintraperitoneal (i.p.) injections of saline or GA (4 or 8 mg/kg) twice (day 0 and day 2). Tumor volume and bodyweight was measured three times a week until the 14thday after the beginning of injection. We found that GA dose-dependently reduced the tumor size (Figs. 1e, f)without inducing any significant loss of body weight(Supplementary Fig. 2). Hematoxylin and eosin (H&E)staining showed that cellular vacuolation was present inMDA-MB 435S xenograft sectionsobtained from GA-treated mice (Fig. 1g). Collectively, these resultsindicate that GA demonstrates an anticancer effect via theinduction of cell death accompanied by vacuolation bothin vitro and in vivo. GA induces paraptosis in cancer cells To investigate which cell death pathway is criticallyinvolved in the anticancer effect of GA, we usedz-VAD-fmk (an apoptosis inhibitor), necrostatin-1 (a necroptosisinhibitor), 3-methyladenine (an early-phase autophagyinhibitor) and bafilomycin A1 (a late-phase autophagyinhibitor). However, the tested inhibitors did not sig-nificantly affect the GA-induced cell death and vacuola-tion of various cancer cell lines (Supplementary Fig. 3),suggesting that apoptosis, necroptosis, and autophagymay not play critical roles in the GA-induced anticancereffect in these cancer cells. To further evaluate the cell death mode induced by GAin our tested cancer cells, we examined the origins of theGA-induced vacuoles. Since autophagy inhibitors did notaffect GA-induced vacuolation, we investigated the pos-sible involvement of mitochondria and/or the ER in thisprocess. Experiments using the YFP-ER plasmid, whichlabels the ER lumen, and the GFP-Sec61β plasmid, whichlabelstthe ER membrane, showed that reticular ERstructures were observed in cells treated with GA for 4 h(Fig. 2a). In contrast, we observed ring-shaped fluores-cence in both YFP-ER (within the ER lumen) and GFP-Sec61β (around the ER vacuole) cells at 8h or GA treat-ment. Thereafter, the dilated ER progressively fused untilmost of cellular spaces (except the nucleus) were occupiedby dramatically expanded ER vacuoles. Analysis employ-ing the YFP-Mito plasmid, which labels the mitochondrialmatrix, revealed that elongated and filamentous mito-chondrial morphologies were detected in untreated cells(Fig.2b). In contrast, large mitochondria-derived vacuoleswere observed around the nuclei of cells treated with 1uM GA for 4 h, and slightly smaller, but still enlargedmitochondria were observed at later time points. Ourtime-lapse imaging revealed that GA initially triggeredmitochondrial swelling, which was followed by the fusionof swollen mitochondria, leading to the formation of giantmitochondria (megamitochondria) with oval or sphericalshape (Fig. 2c). Immunocytochemistry showed that theexpression of succinate dehydrogenase (SDHA, an innermitochondrial membrane protein) was detected as a smalland filled ring shape at the perinuclear area, while theexpression of protein disulfide isomerase (PDI, an ER-resident protein) was observed as a larger ring shape atthe cellular periphery in MDA-MB 435S cells (Fig. 2d). Incontrast, we did not observe any noticeable alteration inthe mitochondria or ER of MCF-10A cells treated withGA. Electron microscopy further revealed that mega-mitochondria and slightly expanded ER structures were observed in MDA-MB 435S cells treated with 2 uM GAfor 8 h, whereas lengthy and filamentous mitochondriaand regular ER structures were detected in untreated cells(Fig. 2e). At 16 h of GA treatment, most of the cellularspaces were occupied by ER-derived vacuoles undergoingfusion. Taken together, these results indicate that GAinduces the morphological features of paraptosis, a celldeath mode accompanied by dilatation of mitochondriaand the ER. Accordingly, we examined whether GA induces thebiochemical features of paraptosis. Although the mole-cular basis of paraptosis still remains to be clarified, it isknown to require de novo protein synthesis° and to becommonly characterized by the induction of ER stress°.We tested the effect of pretreatment with the proteinsynthesis blocker, cycloheximide (CHX), and found that itvery effectively blocked GA-induced cell death andvacuolation in all of the tested cancer cell lines (Figs. 3a,b). The GA-induced mitochondrial dilation observed inYFP-Mito cells and the ER dilation observed in YFP-ERcells were also markedly blocked by CHX pretreatment(Fig. 3c). Since MAP kinases, including ERKs and JNKs,have been positively associated with paraptosis7-9,11,12, wenext investigated whether GA activates these kinases. Incells treated with 1 uMGA, the phosphorylation levels ofERKs and JNKs were markedly increased (Fig. 3d). Inaddition, pretreatment of MDA-MB 435S and MDA-MB453 cells with either PD98059, a MEK inhibitor, orSP600125, a JNK inhibitor, partially but significantlyattenuated GA-induced cell death and vacuolation(Figs. 3e, f). Taken together, these results indicate thatGA-induced cell death in the tested cancer cells shares thebiochemical features of paraptosis. GA induces ER stress due to proteasomal inhibition andtriggers mitochondrial depolarization without ROSgeneration We previously showed that proteasome inhibition-triggered proteostatic disruption critically contributes toparaptosis, particularly in the contexts of ER stress and ERdilation8,9,12. Thus, to investigate the mechanism under-lying GA-induced paraptosis, we tested whether GAaffects proteasome activity. We found that GA treatmentprogressively increased the levels of poly-ubiquitinatedproteins,proteasome-substrate proteins (e.g., Nrfl, Mcl-1,and Noxa) and ER stress marker proteins (e.g., phospho-eIF2a, ATF4, and CHOP) (Fig. 4a). CHX pretreatmenteffectively blocked the GA-induced accumulation of thetested proteasome-substrate proteins and ER stress mar-ker proteins (Fig.4a). Taken together, these results indi-cate that GA induces paraptosis in various cancer cells,and that proteasome inhibition may be closely associatedwith the ER stress and ER dilation seen during thisprocess. Fig.2 GA induces the paraptotic morphologies in cancer cells. a, b YFP-ER or GFP-Sec61B cells (a) and YFP-Mito cells (b) and treated with 1 uMGA for the indicated time points were observed under the confocal microscope. Representative pictures of cells are shown. Bars, 20 um. c Time-lapseimaging results of YFP-Mito cells treated with GA under the confocal microscope. Representative pictures of cells are shown. Bars, 20 um. d Cells weretreated with 1 uM GA for 12 h, fixed, and subjected to the immunocytochemistry of PDI and SDHA. Bars, 20 um. e Transmission electron microscopyof MDA-MB 435S cells treated with 1 uM GA for 24 h. Bars,2 um To investigate how GA affects mitochondrial functionduring paraptosis, we analyzed the mitochondrial mem-brane potential (MMP, Ay) using tetramethylrhodaminemethyl ester (TMRM). Interestingly, although the MMPwas not markedly altered in the megamitochondria ofYFP-Mito cells treated with GA for 4 h, it was lost in thosesubsequently undergoing mitochondrial fragmentation(Figs. 4b, c and Supplementary Fig. 4). Staining of YFP-ER cells with MitoTracker-Red (MTR) showed that thisMMP loss was accompanied by a marked expansion of theER-derived vacuoles (Fig. 4d). The loss of MMP afterexposure to various stimuli is often followed by the pro-duction of reactive oxygen species (ROS), and severalstudies showed that GA-induced ROS play a critical rolein its anticancer effects2,18. Therefore, we tested the pos-sible involvement of ROS in GA-induced paraptosis. Fig. 3 GA induces biochemical features of paraptosis in cancer cells. a Cells pretreated with cycloheximide (CHX) were further treated with GA atthe indicated concentrations for 24 h. Cellular viability was assessed using IncuCyte. Data represent the means ± SEM (n= 3). Statistical significancewas determined using one-way ANOVA followed by Bonferroni's post hoc tests. *p<0.01 vs. untreated control, #p<0.05 vs. GA-treated cells. b Cellspretreated with the 2 uM CHX were further treated with GA (0.5 uM for SNU-668; 1 uM for MDA-MB 435S, BxPC-3 and SNU-449; 2 uM for MDA-MB453; 3 uM for MDA-MB 468 and NCI-H460 cells) for 12 h, and observed by the phase-contrast microscopy. Bars, 40 um. c YFP-Mito or YFP-ER cellspretreated with 2 uM CHX and further treated with 1 uM GA for 8 h were observed under the confocal microscope. Bars, 20 um. d MDA-MB 435S cellswere treated with 1 uM GA for the indicated time points. Western blotting of the indicated proteins was performed. B-actin was used as a loadingcontrol.e Cells were pretreated with PD98059 (PD) or SP600125 (SP) and further treated with GA at the indicated concentrations for 24 h. Cellularviability was assessed using IncuCyte. Data represent the means±SEM (n=3). Statistical significance was determined using one-way ANOVAfollowed by Bonferroni's post hoc tests. *p<0.01 vs. untreated control, #p<0.05 vs. GA-treated cells. FCells pretreated with the 20 uM PD or SP werefurther treated with GA (1 uM for MDA-MB 435S and 2 uM for MDA-MB 453) for 12 h and observed by the phase-contrast microscopy. Bars, 40 um However, measurement of ROS using CM-HDCF-DArevealed that GA treatment did not noticeably increasethe levels of ROS in the tested cancer cells, whereas HO2treatment (positive control) did trigger such a response(Fig. 4e and Supplementary Fig.5). Thiol-containing antioxidants effectively block GA-inducedparaptosis, independently of ROS generation To confirm that ROS are not involved in GA-inducedparaptosis, we tested the effects of various antioxidants.Interestingly, the general antioxidant, N-acetylcysteine(NAC), but not the mitochondria-targeted antioxidant,Tiron, dose-dependently blocked GA-induced cell deathand vacuolation in all of the tested cancer cell lines(Figs. 5a, b). When we further examined the effects ofother antioxidants on GA-induced cellular responses, wefound that thiol-containing antioxidants, including glu-tathione (GSH), and N-(2-mercapto-propionyl)-glycine(NMPG), but not non-thiol ROS scavengers, such as,ascorbic acid (Vitamin C) and manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP; a superoxidedismutase mimetic), also very effectively blocked GA-induced cell death in all of the tested cancer cells (Fig.5c).Moreover, the GA-induced cellular vacuolation in thesecells (Fig. 5d), the dilation of the ER in YFP-ER cells andthe dilation of mitochondria in YFP-Mito cells (Fig. 5e)were effectively blocked by various thiol antioxidants butnot by non-thiol antioxidants. Collectively, these resultssuggest that thiol-antioxidants block GA-induced para-ptosis in a ROS-independent manner. The formation of an adduct between GA and thiol-containing proteins may critically contribute to GA-induced paraptosis Considering the highly electrophilic character of the a,B-unsaturated ketone substructure of GA at C1020.21, weexamined the possibility that GA may react with the thiolgroups of GSH and NAC to form covalent Michaeladducts, such as GA-GSH and GA-NAC conjugates(Fig. 6a). To test this hypothesis, we incubated 100 uMGA with excess 50 mM GSH for 3 h and then analyzed thereaction mixture by LC-MS/MS.The MS scan revealed apeak at m/z 936 ([M+H]+), which corresponded to the molecular weight of the Michael addition product, and itsrepresentative fragmentation pattern in the product ionscan showed peaks at m/z 629 ([M+H]+) and 308 ([M+H]+), which corresponded to the molecular weights of GAand GSH, respectively (Fig. 6b). The LC-MS/MS chro-matogram obtained after incubation of 100 uM GA with50 mM NAC showed the GA-NAC adduct peak at m/z814 ([M+Na]*), confirming that an adduct was formedbetween GA and NAC. To further examine whether theinteraction between GA and NAC affected GA-mediatedcytotoxicity, we pre-incubated cells with 1 pM GA plusdifferent doses of NAC in serum-free medium at roomtemperature for different durations to allow the formationof chemical adducts, and then treated MDA-MB 435Scells with each GA-NAC mixture for 24 h. At a given doseof NAC, cells treated with GA and NAC that hadundergone the prolonged pre-incubation showed far lessGA-mediated cytotoxicity than those subjected to simul-taneous treatment; moreover, pre-incubation required alower concentration of NAC to block GA-mediated celldeath to the same extent, compared to simultaneoustreatment (Fig. 6c). These results strongly suggest thatNAC blocks GA-induced cytotoxicity by eliminating itsability to form Michael adducts, particularly with thenucleophilic thiol groups of intracellular proteins. Tofurther test whether GA directly reacts with the free thiolresidues of proteins, we performed the dibromobimane(dBrB) assay, which is based on the ability of dBrB to reactwith free reduced thiols and generate a highly fluorescentprotein-dBrB adduct22,23. We used iodoacetamide (IAM),an alkylating agent that reacts with protein-SH groups toform stable S-carboxyaminodimethyl-cysteineadducts23,24, as a positive control. Indeed,IAM treat-ment effectively reduced the free protein-SH levels inMDA-MB 435S cells (Fig. 6d). Importantly, GA treatmentalso dose-dependently decreased the protein-SH levels inthese cells, suggesting that stable adducts formed betweenGA and thiol-containing proteins to disrupt intracellularthiol homeostasis. Supporting this idea, the GA-inducedaccumulations of poly-ubiquitinated proteins, phospho-eIF2c, ATF4 and CHOP were effectively inhibited only bythiol antioxidants (Fig. 6e). In addition, the GA-inducedloss of MMP was almost completely blocked by NAC Fig. 4 ROS levels are not noticeably increased in spite of the MMP loss in GA-induced paraptosis. a MDA-MB 435S cells pretreated with orwithout 2 uM CHX and further treated with 1 uM GA for the indicated time points. Cell extracts were prepared for Western blotting of the indicatedproteins. Western blotting of B-actin was used as a loading control. b,c YFP-Mito cells (b) or MDA-MB 435S cells (c) were treated with 1 uM GA forthe indicated time points or 20 uM CCCP for 12 h were incubated with TMRM. Samples were subjected for confocal microscopy (b) or flow cytometry(c). d YFP-ER cells were treated with 1 uM GA for the indicated time points or 20 uM CCCP for 12 h. Treated cells were incubated with MTR andobserved under the confocal microscope. e Cells treated with GA (1 uM for MDA-MB 435S; 2 uM for MDA-MB 453; 3uM for MDA-MB 468 and NCI-H460 cells) for 16h or 5 mM H0, for 10 min were incubated with CM-H,DCF-DA (DCF-DA) and subjected for the fluorescence microscopy. Bars,40 um treatment (Fig. 6f). Taken together, our results suggestthat the GA-induced covalent modification of the freethiol groups of intracellular proteins may interfere withproper disulfide bond formation during protein folding andinduce the accumulation of misfolded proteins withinthe ER and mitochondria, leading to stress and dilation ofthese organelles, and eventual11paraptotic cell death(Fig.7). Discussion GA has been shown to demonstrate anticancer effectsby inhibiting the cell growth of various types of humancancer cells in vitro and in vivo3,25-28, by inducingapoptosis3,25,26,29 ind by inhibitingmetastasiss andangiogenesis30.31. Several in vivo studies on adults of dif-ferent animal species have demonstrated that GA exhibitsa good safety profile. It has also been shown to enhancethe cytotoxic effects of various chemotherapeutic agents,such as docetaxel33, oxaliplatin34, adriamycin3, imatinib,and bortezomib,in various cancer cells, while reducingthe side effects associated with these agents. Thus, GAappears to have great potential as an effective agent forthe preventionaand treatment of different cancers. However, despite its potent anticancer efficacy, themolecular mechanisms underlying the actions of GAremain still elusive. In this study, we show for the firsttime that GA can induce antitumor effects by inducingparaptosis as a major cell death mode. In the tested cancercells, GA treatment rarely yielded apoptotic morphologiesand the GA-induced anticancer effect was not sig-nificantly dependent on caspases. Given the problemswith malignant cancer cells showing genetic heterogeneity37,38as well as innate and acquired resistance to apoptosisit may be useful to apply a strategy to induce an alter-nativeeCcancer cell deathmodeewhoseregulatorymechanisms differ from those of apoptosis. Paraptosis issuch a cell death mode, and its induction may greatly Fig. 5 (See legend on next page.) and further treated with GA at the indicated concentrations for 24 h. Cellular viability was assessed using IncuCyte. Data represent the means ± SEM(n=3). Statistical significance was determined using one-way ANOVA followed by Bonferroni's post hoc tests. *p<0.01 vs. untreated control, #p<0.05 vs. GA-treated cells. b, d Cells were pretreated with 2 mM NAC or 1 mM Tiron and further treated with GA (0.5 pM for SNU-668; 1 pM for MDA-MB435S, BxPC-3 and SNU-449; 2 uM for MDA-MB 453; 3 pM for MDA-MB 468 and NCI-H460 cells) for 12 h. Cells were observed by the phase-contrastmicroscopy. Bars,40 um. e YFP-Mito or YFP-ER cells pretreated with 2 mM NAC, 2 mM GSH, 0.4 mM NMPG, 1 mM Tiron, 0.25 mM ascorbic acid (A.A) or100 uM MnTBAP and further treated with 1 uM GA for 8 h were observed under the confocal microscope. Bars, 20 um contribute to improving cancer therapy. We observedextensive vacuolation and subsequent cell death in varioustypes of cancer cells exposed to GA and in MDA-MB435S xenograft sections obtained from GA-treated mice.This GA-induced vacuolation originated from the ER andmitochondria, asassessed using relevant expressionplasmids (YFP-ER, GFP-Sec61β and YFP-Mito), immu-nocytochemical analysis of changes in the ER or mito-chondria, and electron microscopy. In response to GA, megamitochondria formed due to the swelling and fusionof mitochondria; this was followed by the expansion ofER-fusion-derived vacuoles. Pretreatment with CHX,which is known as an inhibitor of paraptosis, veryeffectively blocked the dilation of the ER and mitochon-dria and subsequent cell death in the tested cancer celllines. If we hope to deconvolute the mechanisms of actionunderlying the biological or therapeutic action of GA, we must understand its molecular targets. GA has beenshown to directly bind and inhibit various functionallyimportant proteins,including proteasome ,theHsp90p IKKB4,, topoisomeraseIIa42,, transferrinreceptor43, thioredoxin 1/244, MDM24, and XPO24.However, the primary cellular target(s) and action mode (s) of GA remain unclear. It is also interesting to considerhow GA can act as an effective multi-targeted agent. The9,10-carbon double bond of the a,B-unsaturated ketone ofGAwasproposed to be imperative for its cytotoxi-city20,21,29and has been shown to directly interact withthe cysteine residues of IKK4, thioredoxin 1/24, and (see figure on previous page)Fig.6 The activity of GA to bind to thiol-containing proteins may be critical for its paraptosis-induced ability in cancer cells. a Proposedchemical structures of the GA-GSH and GA-NAC adducts. b Full-scan product ion scan spectra and the expected structures of GA, GA-GSH, and GA-NAC adduct formed upon Michael addition of GSH or NAC. The m/z values of the GA-GSH adduct represent GSH at 308, GA at 629, and the adductform at 936. The m/z values of the GA-NAC adduct represent NAC at 164, GA at 651, and the adduct form at 814. c Increasing concentrations of NACwere pre-incubated with 1 pM GA in serum-free medium for the indicated time durations at room temperature, and these mixtures were used totreat MDA-MB 435S cells for 24h. The cell viability was measured using IncuCyte. Data represent the means±SD. Kruskal-Wallis test was performedfollowed by Dunn's test. *p <0.05; **p<0.01 vs. GA-treated cells. d MDA-MB 435S cells were treated with the indicated concentrations of GA or10 pM iodoacetamide (IAM; a positive control to reduce intracellular protein-SH levels) for 12 h. Protein-SH levels were measured using thedibromobimane (dBrB) assay, as described in the Materials and Methods. Data represent the means ±SD. Kruskal-Wallis test was performed followedby Dunn’s test. *p<0.01 vs. untreated control. e Cell extracts were prepared from the cells pretreated with the antioxidants and further treated withGA (1 uM for MDA-MB 435S and 2 uM for MDA-MB 453 cells) for the indicated concentrations for Western blotting. B-actin was used as a loadingcontrol. f MDA-MB 435S cells pretreated with 2 mM NAC and further treated with 1 uM GA for the indicated time points were incubated with TMRM.Samples were subjected for flow cytometry Fig. 7 Hypothetical model for the underlying mechanisms of GA-induced paraptosis XPO24. Here, we propose that the 9,10-carbon doublebond of the a,p-unsaturated ketone of GA may act as anelectrophilic Michael addition center to covalently modifynucleophilic cysteinyl thiols of its multiple target proteinsto trigger paraptosis. This hypothesis is supported by thefollowing findings presented herein: (a) GA undergoeschemical reactions with GSH and NAC via their thiolnucleophilic thiol structures to generate GA-GSH andGA-NAC adducts, respectively, in vitro. ((b) Pre-incubation of NAC with GA ameliorates the cytotoxicityof GA toward MBA-MB 435S cells. (c) Various thiolantioxidants block all of the tested GA-induced cellular responses, including proteasome inhibition, ER stress, ERdilation, mitochondrial dilation and MMP loss,leading tocell death in cancer cells of different tissue origins. (d) GAtreatment reduces the intracellular sulfhydryl protein(protein-SH) content in MDA-MB 435S cells, suggestingthat the formation of Michael adducts with intracellularcysteinyl thiols of proteins may critically contribute toGA-induced toxicity. Given this, we might next ask: Whatis the underlying mechanism by which GA triggers dila-tion of the ER and mitochondria? One possible mechan-ism is that the electrophilic nature of GA may disruptproper disulfide bond formation to cause misfolding ofthe most accessible proteins, leading to accumulation ofmisfolded proteins within the ER and mitochondria. Inparticular, GA-mediated binding and subsequent inhibi-tion of the machineries associated with proteostasis, suchas the proteasome, may result in a dramatic accumulationof misfolded proteins in the cell. Previously, Mimnaughet a1.47proposed that a proteasome-inhibition-triggeredoverload of misfolded proteins in the ER lumen couldexert an osmotic pressure that draws water from thecytoplasm to distend the ER lumen. In the present study,we found that GA inhibited proteasome activity, as evi-denced by the accumulation of poly-ubiquitinated pro-teins and proteasome-substrate proteins. Inhibition of denovo protein synthesis by CHX pretreatment blocked thisGA-mediated response, possibly reducing the protein loadin the ER. Accumulation of misfolded proteins within themitochondria has also been proposed to lead to mito-chondrial stress and dilation48,49, in a manner similar toER dilation. However, the exact cause of mitochondrialdilation remains to be clarified. Ultimately, the physicaland functional perturbation of the two organelles maylead to MMP loss and irreversible paraptotic cell death.Regarding the potential targets through which GA couldcontribute to paraptosis, we speculated that MDM2 maybe involved. GA has been reported as a putative MDMinhibitor and a fraction of MDM2 was recently shown tobe imported to control mitochondrial dynamics inde-pendently of p53. In addition, we recently reported that the MDM2 inhibitor, nutlin-3, induces mitochondrialdilation49.Furthermore, combination treatment of MDA-MB 435S cells with nutlin-3 and proteasome inhibitors(e.g., bortezomib) effectively induced paraptotic celldeath. Based on the present and previous results, wehypothesized that GA may induce paraptosis via dualinhibition of the proteasome and MDM2.To address this,we first tested whether GA-mediated paraptosis can besensitized by inhibition of the proteasome or MDM2. Wefound that low doses of GA (up to 0.5uM), bortezomib(up to 10nM) or nutlin-3 (up to 40 uM) did not notice-ably induce cellular vacuolation and cell death (Supple-mentary Fig. 6A,B). However, bortezomib or nutlin-3dose-dependently enhanced vacuolation and cell death inMDA-MB 435S cells treated with 0.5 uM GA, similar tothe effect of bortezomib plus nutlin-3 (SupplementaryFig.6A, B). In addition, 5 nM bortezomib plus 0.5 uM GAor 30 uM nutlin-3 plus 0.5 uM GA markedly induced thedilation of both the ER and mitochondria, similar to theeffect of bortezomib plus nutlin-3 (SupplementaryFig.6C). These observations begin to suggest that GA mayinhibit both the proteasome and MDM2, although theseactivities should be further validated. A a,B-unsaturatedketone structure was proposed to be able to target andinhibit the proteasome chymotrypsin-like 5 subunit,and a,B-unsaturated ketone group of GA can form acovalent bond with a nucleophilic residue by the Michaelreaction.Therefore,, we tested the possible covalentdocking between GA and the SH groups of cysteineresidues in this proteasome subunit using molecularmodeling studies. Indeed, our computational dockingstudies supported the potential of C10 atom of GA tobind to the Cys52 residue of the proteasome 20Sβ5 subunit (Supplementary Fig. 7A). In addition, we foundthat the C10 atom of GA demonstrates possible covalentbinding to the Cys77 residue of MDM2 (SupplementaryFig. 7B). Thus, our molecular modeling studies suggestthat GA can bind covalently with cysteine residues of theproteasome or MDM2 and thus, the proteasome andMDM2 may be included as potential targets of GA for theinduction of paraptosis. Further detailed studies will beneeded to test this hypothesis. To selectively kill cancer cells, it is advantageous toexploit the differential characters of cancer cells andnormal cells. Tumor cells often suffer from higher ERstress than normal cells due to the imbalance between ahigh metabolic demand and a limited protein-foldingcapacity. When we examined whether GA-induced ERstress is differently modulated between breast cancer cellsand normal cells, we found that GA treatment increasedthe levels of eIF2x phosphorylation and CHOP protein inMDA-MB 435S cells at its lower concentrations, com-pared to those in MCF-10A cells treated with GA (Sup-plementary Fig. 8A). Noxa upregulation was also more potently induced in GA-treated MDA-MB 435S cells,compared to that in GA-treated MCF-10A cells. Addi-tionally, cancer cells exhibit greater oxidative stress thannormal cells due to oncogenic stimulation, increasedmetabolic activity and mitochondrial malfunction . Toinvestigate the GA-induced functional change in breastcancer cells and normal cells, we analyzed the MMP usingMTR together with Acridine Orange 10-Nonyl Bromide(NAO), a mitochondrial specific fluorescent probe. MMPloss was observed in MDA-MB 435S cells treated with GAfor 16 h, but not in GA-treated MCF-10A cells (Supple-mentary Fig. 8B). These results suggest that GA mayconfer cancer cells much more stress to the ER andmitochondria than normal cells. In this respect, GA-mediated paraptosis may provide a two-pronged attack asa therapeutic strategy for selectively killing cancer cellsthat are already under the stress inherent to cancer. Materials and methods Chemicals and antibodies Gambogic acid (GA) was purchased from Biorbyt (SanFrancisco, CA, UK). Manganese (III) tetrakis (4-benzoicacid) porphyrin chloride (MnTBAP) was from Calbio-chem (EMD Millipore Corp., Billerica, MA,,USA).MitoTracker-Red (MTR), propidium iodide (PI), 5-(and-6)-chloromethyl-2',7'-dichlorofluorescein diacetate (CM-H2DCF-DA), and tetramethylrhodamine methyl ester(TMRM) along with secondary antibodies (rabbit IgGHRP (G-21234) and mouse IgG HRP (G-21040)) werefrom Molecular Probes (Eugene, OR, USA). The primaryantibody against SDHA(ab14715) was from Abcam(Cambridge, MA, USA) and Noxa (OP180) was fromCalbiochem. B-actin (sc-47778), ubiquitin (sc-8017), Mcl-1 (sc-819), ATF4 (sc-200) were from Santa Cruz Bio-technology (Santa Cruz, CA, USA). Phospho-SAPK/JNK(#9251), SAPK/JNK (#9252), phospho-ERK1/2 (#9101),ERK1/2 (#9102), TCF11/NRF1 (#8052), CHOP/GADD153 (#2895), phospho-eIF2α (#9721), and eIF2a(#9722) were from Cell Signaling Technology (Danvers,MA, USA). PDI (ADI-SPA-890-F) was form Enzo LifeScience (Farmingdale, NY, USA). Other reagents werefrom Sigma-Aldrich (St Louis, MO, USA). Cell culture MDA-MB 435S, MDA-MB 453,MDA-MB 468, MCF-10A, BxPC-3, and NCI-H460 cells were purchased fromAmerican Type Culture Collection (ATCC, Manassas,VA, USA). SNU-449 and SNU-668 cells were purchasedfrom Korean Cell Line Bank (KCLB, Seoul, Korea). MDA-MB 435S cells were cultured in DMEM and MDA-MB453, MDA-MB 468, BxPC-3, NCI-H460, SNU-449 andSNU-668 cells were cultured in RPMI-1640 mediumsupplemented with 10% fetal bovine serum and 1% anti-biotics (GIBCO-BRL, Grand Island,NY,USA). MCF-10A cells were maintained in Mammary Epithelial GrowthMedium (MEGM; Clonetics Corp., San Diego, CA, USA)supplemented with pituitary extract, insulin, human epi-dermal growth factor, hydrocortisone and choleratoxin(Calbiochem). Cells were incubated in 5% CO2 at 37℃. Cell viability assay Cells were cultured in 48-well plates and treated asindicated. The cells were then fixed with methanol/acet-one (1:1) at -20℃ for 5 min, washed three times withPBS and stained with propidium iodide (PI; final con-centration,1 ug/ml) at room temperature for 10 min. Theplatess were imaged onanIncuCyte device (EssenBioscience, Ann Arbor, MI, USA) and analyzed using theIncuCyte ZOOM 2016B software. The processing defini-tion of the IncuCyte program was set to recognizeattached (live) cells by their red-stained nuclei. The per-centage of live cells was normalized to that of untreatedcontrol cells (100%). In vivo tumor growth inhibition assay MDA-MB 435S cells were used to produce a xenografttumor model in female BALB/c nude mice (nu/nu, 5 weeksold, Japan SLC, Hamamatsu, Japan). A suspension of 5×10°cells in a 50 ul volume (saline) was subcutaneously injectedinto the right flank of mice.Tumors were grown for 3 weeksuntil average tumor volume reached 100~150 mm. Micewere randomized into 3 groups (n=4 per group), includingvehicle (PBS containing 0.25% DMSO), 4mg/kg GA and8 mg/kg GA, and mice were received twice intraperitoneal(i.p.) injections of GA at the indicated concentrations (day 0and day 2). Tumor size was measured three times a week for2 weeks and tumor volume was calculated using the formula[V=(Lx W2) x 0.5, where V=volume, L=length, andW=width]. All experiments were performed following theguidelines and regulations approved by the InstitutionalAnimal Care and Use Committee of the Asan Institute forLife Science. On the 14th day, mice were sacrificed and thetumors were isolated, fixed in 4% paraformaldehyde andthen embedded into paraffin. Sections of 5 um were stainedwith H&E and the image on the tissue sections was observedand photographed by CMOS (Complementary metal-oxide-semiconductor) camera which is attached on K1-Fluomicroscope (Nanoscope Systems, Daejeon, Korea). Examination of the morphologies of mitochondria and theER employing the plasmids to specifically label the ER ormitochondria Establishment of the stable cell lines expressing thefluorescence specifically in the ER lumen (YFP-ER cells)and the cell lines expressing the fluorescence specificallyin mitochondria(YFP-Mito cells) werepreviouslydescribed55. Additionally, to label the ER membrane,MDA-MB 435S cells were transfected with the GFP- Sec61β (Addgene plasmid #15108) and the stable cell lineswere selected with medium containing 500 ug/ml G418(Calbiochem). Morphological changes of mitochondria orthe ER were observed under confocal laser scanningmicroscope (K1-Fluo) using filter set (excitation bandpass, 488 nm; emission band pass,525/50). Immunoblot analyses and immunofluorescencemicroscopy Immunoblot and immunofluorescence analysiswasperformed as described previously. Images were acquiredfrom Axiovert 200M fluorescence microscope (CarlZeiss, Oberkochen, Germany) using Zeiss filter sets #46(excitation band pass, 500/20nm; emission band pass,535/30 nm), and #64HE (excitation band pass, 598/25 nm;emission band pass,647/70nm). Transmission electron microscopy Cells were prefixed in Karnovsky's solution (1% paraf-ormaldehyde, 2% glutaraldehyde, 2 mM calcium chloride,0.1 M cacodylate buffer, pH 7.4) for 2 h and washed withcacodylate buffer. Post-fixing was carried out in 1%osmium tetroxide and 1.5% potassium ferrocyanide for1 h. After dehydration with 50-100% alcohol, the cellswere embedded in Poly/Bed 812 resin (Pelco, Redding,CA), polymerized, and observed under electron micro-scope (EM 902 A, Carl Zeiss). Measurement of ROS generation Treated cells were incubated with 10 uM of CM-HDCF-DA for 30 min at 37℃, and subjected for thefluorescence microscopy. Images were acquired fromAxiovert 200 M fluorescence microscope using Zeiss filtersets #46 (excitation band pass, 500/20 nm; emission bandpass, 535/30 nm). Analysis of mitochondrial membrane potential To analyze mitochondrial membrane potential (Ay),cells (8×10 cells) cultured in 12-well plates were treatedwith 1uM GA or 20uM carbonyl cyanide m-chlor-ophenyl hydrazine (CCCP) for the indicated time pointsand incubated for 30 min at 37°C with 200 nM TMRM or100 nM MTR. After washingwith PBS, cells; wereobserved under confocal laser scanning microscope (K1-Fluo). For fluorescence-activated cell sorting analysis,TMRM stained cells were washed PBS, detached fromdishes, and subjected for using a FACScan system (BDBiosciences, SanJose, CA, USA). In vitro reactions of GA with GSH or NAC, and LC-MS/MSanalysis GA was adjusted to 100 uM in 0.1 mL methanol andthen mixed with 0.1 mL of either 50 mM GSH or 50 mMNAC. After 3h incubation at 40C, the reaction was quenched by the addition of 20-fold ice-cold methanol.For monitoring of GA alone and its formation of adductswith NAC or GSH, 1-uL aliquot of the mixture wasdirectly injected into an Agilent 6470 Triple Quad LC-MS/MS system (Agilent, Wilmington, DE, USA) coupledto an Agilent 1260 HPLC system. Chromatographicseparation was performed on a Synergi Polar RP (4um,2.0 mm i.d. x150 mm, Phenomenex, Torrance, CA,USA)using a mobile phase that comprised water and methanol(15:85, v/v) containing 0.1% formic acid. Mass spectrawere recorded by electrospray ionization in the positivemode. Detoxification of GA with N-acetylcysteine (NAC) To test the detoxification of GA by NAC, aliquots ofserum-free DMEM containing 1 uM GA and differentconcentrations of NAC were pre-incubated at roomtemperature for the indicated time points, and thenincubated with MDA-MB 435S cell cultures for 24 h. Toexamine the effect of simultaneous treatment of GA andNAC, cells were treated with 1 uM GA and increasingconcentrations of NAC without pre-incubation. Subse-quently, cell viability was assessed by PI staining usingIncuCyte. Fluorescence labeling of protein thiol groups bydibromobimane(dBrB) assay MDA-MB 435S cells plated in 60 mm plates weretreated as indicated, harvested, resuspended in PBS, andsonicated. A part of samples was used for the measure-ment of the protein concentration using Lowry-basedassay and the rest of samples were immediately reactedwith 1.5 N perchloric acid and incubated for 5 minutes onice to precipitate the proteins. The samples were cen-trifuged at 14,000 x g for 10 minutes, and the pelletedproteins were solubilized with 0.1M NaOH and neu-tralized using 0.5M Tris-HCl. The prepared proteins(1 ug) were mixed with 1 uM dibromobimane and incu-bated for 40 min at 37℃. Dibromobimane-bound pro-tein-SHgroupsswWeremeasured ina fluorescencemultiplat:e reader (Synergy H1 HybridMulti-Modereader; BioTek, Winooski, VT, USA) at Ex/Em 393/477nm. Fluorescence was normalized by the total proteinlevels and expressed as percentage of protein-SH levelscompared to that from the untreated group. Statistical analysis All data are presented as mean ±SEM (standard error ofthe mean) or±SD (standard deviation) from at least threeseparate experiments. To perform statistical analysis,GraphPad Prism (GraphPad Software Inc, Sandiego, CA)was used.Normalityoffddata Was assessedbyKolmogorov-Smirnov testes and equal variance usingBartlett’s test. For normal distribution, statistical differences were determined using an analysis of variance (ANOVA)followed by followed by Bonferroni multiple comparisontest.Ifthedata were notnormally distributed,Kruskal-Wallis test was performed followed by Dunn’s test. Acknowledgements This work was supported by the grants from the Korean Health TechnologyR&D Project, Ministry of Health & Welfare (HI18C0263). Author details 'Department of Biomedical Sciences, Ajou University Graduate School ofMedicine, Suwon 16499, Korea. Department of Biochemistry and MolecularBiology, Ajou University, Suwon 16499, Korea.Department of Pharmacy, AjouUniversity, Suwon 16499, Korea. “Department of Pharmacy, DankookUniversity, Cheonan 16890, Korea. Chemical Data-Driven Research Center,Korea Research Institute of Chemical Technology, Daejeon 34114,Korea. AsanInstitute for Life Sciences, Department of Convergence Medicine, Asan MedicalCenter, University of Ulsan Collegeof Medicine, Seoul 05505, Korea.Center forAdvancing Cancer Therapeutics, Department of Radiation Oncology, AsanMedical Center, University of Ulsan College of Medicine, Seoul 05505, Korea Conflict of interest The authors declare that they have no conflict of interest Publisher's note Springer Nature remains neutral with regard to jurisdictional claims inpublished maps and institutional affiliations. Received: 5 September 2018 Revised: 25 December 2018 Accepted: 9January 2019 Published online: 22 February 2019 ( References ) ( 1. Yang, L . J . & Chen, Y. New targe t s for th e antitumor activity of gambogic acidin hematologic m a lignancies. Acta Pharmacol. Sin. 34, 191-198 (2013). ) ( 2. Banik, K. et al. Therapeutic potential of gambogic acid, a caged xanthone, totarget cancer. Cancer Lett. 4 16, 7 5 -8 6 (2018). ) 3.Shi, X. et al. Gambogic acid induces apoptosis in Imatinib-resistantchronic myeloid leukemia cells via inducing proteasome inhibition andcaspase-dependent Bcr-Abl downregulation. Clin. Cancer Res 20, 151-163(2014). 4.Yang, Y.et al. Differential apoptotic induction of gambogic acid, a novelanticancer natural product, on hepatoma cells and normal hepatocytesCancer Lett. 256, 259-266(2007). 5.Chi, T. et al. An open-labeled, randomized, multicenter phase lla study ofgambogic acid injection for advanced malignant tumors. Chin. Med. J. (Engl.)126, 1642-1646 (2013). 6.Sperandio,S.,de, Belle, 1. & Bredesen, D. E. An alternative, nonapoptotic form ofprogrammed cell death. Proc. Natl. Acad. Sci. USA 97, 14376-14381 (2000). 7. Sperandio, S. et al. Paraptosis: mediation by MAP kinases and inhibition byAIP-1/Alix. Cell. Death. Differ. 11, 1066-1075 (2004) 8.Lee, D., Kim, I. Y., Saha, S. & Choi, K. S. Paraptosis in the anti-cancer arsenal ofnatural products. Pharmacol. Ther. 162, 120-133(2016). ( 9. Y oon, M . J . , Kim, E. H , Lim, J . H . , Kwon, T. K. & Choi , K. S . Superoxide anion andproteasomal dysfunction c ontribute to c u rc u min-induced pa r aptosis ofmalignant breast cancer cells. Free. Radic . Biol. Me d 48 , 713 - 726 (2010). ) 10 Yoon, M. J, Kim, E. H., Kwon, T. K, Park, S. A. & Choi, K. S. Simultaneousmitochondrial Ca+overload and proteasomal inhibition are responsible forthe induction of paraptosis in malignant breast cancer cells. Cancer Lett. 324,197-209 (2012). 11. Yoon, M. J. et al. Release of Ca+ from the endoplasmic reticulum and itssubsequent influx into mitochondria trigger celastrol-induced paraptosis incancer cells. Oncotarget 5, 6816-6831 (2014). ( 1 2 Y oon, M. J. et al. Stronger proteasomal inhibiti o n and higher CHOP inductionare responsible for more effective i nduction of p araptosis by dimethox-ycurcumin than curcumin. C e l l. Death. D i s. 5 , e1112 ( 2014). ) ( 13 Kar, R., Singha, P. K., V enkatachalam, M . A. & Saikum a r, P. A novel role for MAP1LC3 i n n onautophagic cytoplasmic va c uolation de a th of cancer cel l s. Onco- gene 28, 2 556-2568( 2 009). ) ( 14 Singha, P. K., Pandeswara, S., Venkatachalam, M . A. & Saikumar, P. ManumycinA inhibits triple-negative breast cancer growth t h rough LC 3 -mediated cyto - plasmic v acuolation d e ath. C e ll. D eath. D is. 4, e457 (2013). ) ( .1 5 Kim, I . Y. et al. Ophi o bolin A k i ll s human glioblastoma cells by i nducingendoplasmic re t iculum stress via disruption of thiol p r oteostasis. Oncotarget 8, 106740-106752 (2017). ) 16 Park, S. S. et al. Pyrrolidine dithiocarbamate reverses Bcl-xL-mediated apoptoticresistance to doxorubicin by inducing paraptosis. Carcinogenesis 39, 458-470(2018). .7.Henry-Mowatt, J., Dive, C., Martinou,J. C. & James, D. Role of mitochondrialmembrane permeabilization in apoptosis and cancer. Oncogene 23,2850-2860 (2004). 18 Pan, H. et al. Gambogic acid inhibits thioredoxin activity and induces ROS-mediated cell death in castration-resistant prostate cancer. Oncotarget 8,77181-77194(2017). 19. Oyewole, A. O. & Birch-Machin, M. A. Mitochondria-targeted antioxidantsFaseb.J.29, 4766-4771 (2015). 20 Zhang, H. Z. et al. Discovery, characterization and SAR of gambogic acid as apotent apoptosis inducer by a HTS assay. Bioorg. Med. Chem. 12, 309-317(2004). 21 Han, Q. B. et al. Stability and cytotoxicity of gambogic acid and its derivative,gambogoic acid. Biol. Pharm. Bul.28, 2335-2337 (2005). .22 Rudyk, O.& Eaton, P. Biochemical methods for monitoring protein thiol redoxstates in biological systems. Redox Biol. 2, 803-813 (2014). .233. Zanotto-Filho, A. et al. Alkylating agent-induced NRF2 blocks endoplasmicreticulum stress-mediated apoptosis via control of glutathione pools andprotein thiol homeostasis. Mol. Cancer Ther. 15, 3000-3014 (2016). 24 Anderson, P. J. & Perham, R. N. The reactivity of thiol groups and the subunitstructure of aldolase. Biochem. J. 117, 291-298 (1970). ( 25 Shi , X. et al. Gambogic acid induces apoptosis in diffuse large B-cell lymphomacells via inducing p roteasome inhibition. Sc i Rep. 5,9694 (2015). ) .26 Huang, G. M., Sun, Y., Ge, X., Wan, X. & Li, C. B. Gambogic acid inducesapoptosis and inhibits colorectal tumor growth via mitochondrial pathways.World J. Gastroenterol. 21,6194-6205 (2015). 27. Zhao, K. et al. Gambogic acid suppresses cancer invasion and migration byinhibiting TGF口1-induced epithelial-to-mesenchymal transition. Oncotarget 8,27120-27136 (2017). 28 Tang, Q. et al. Gambogic acid inhibits the growth of ovarian cancer tumors byregulating p65 activity. Oncol. Lett. 13, 384-388 (2017). 29 Nie, F. et al. Reactive oxygen species accumulation contributes to gambogicacid-induced apoptosis in human hepatoma SMMC-7721 cells. Toxicology260, 60-67 (2009). ( 30 Yi, T. et al.Gambogic a cid i nhibits angiogenesis and prostate tumor growth bysuppressing vascular e ndothelial g rowth f a ctor receptor 2 s i gnaling. C ancer Res 68, 1 8 43-1850 ( 2 008). ) ( 31 Wang, X . & Chen, W. Gambogic acid is a novel anti-cancer agent tha t inh i bitscell proliferation , angiogenesis and metastasis. Anticancer. A gents Med. Ch e m. 1 2,994- 100 0 (2012). ) ( 32 2. Zhao, L. et al. General pharmacological p r opertie s , developmental toxicity, andanalgesic activit y of gambogic acid, a novel natural anticancer agent. Drug C hem. Toxicol. 33, 88-96 (2010). ) 33 Wang, T. et al. Gambogic acid, a potent inhibitor of survivin, reverses docetaxelresistance in gastric cancer cells. Cancer Lett. 262, 214-222 (2008). ( 34 Wang,L. H . e t al. Gambogic ac i d sy n ergistically potentiates ci s platin-inducedapoptosis i n n on-small-cell lung c ancer t hrough s u ppressing NF-KB and MAPK/HO-1 signaling. Br. J.Cancer 1 1 0, 341 -3 52 ( 2014). ) .35 Wang, L. H. et al. Suppression of NF-kB signaling and P-glycoproteinfunction by gambogic acid synergistically potentiates Adriamycin-inducedapoptosis in lungg cancer. Curr. Cancer Drug.. Tlargets 14, 91-103(2014). .36 Liu, N. et al. The combination of proteasome inhibitors bortezomib andgambogic acid triggers synergistic cytotoxicity in vitro but not in vivo. Toxicol.Lett. 224, 333-340 (2014). 37. Kim, R., Emi, M. & Tanabe, K. The role of apoptosis in cancer cell survival andtherapeutic outcome. Cancer Biol. Ther. 5, 1429-1442 (2006) .38 Mohammad, R. M. et al. Broad targeting of resistance to apoptosis in cancerSemin. Cancer Biol. 35, S78-S103 (2015). 39 Li,X. et al. Gambogic acid is a tissue-specific proteasome inhibitor in vitro andin vivo. Cell. Rep. 3,211-222(2013). .40 Yim, K. H. et al.Gambogic acid identifies an isoform-specific druggable pocketin the middle domain of Hsp90B. Proc. Natl. Acad. Sci. USA 113, E4801-E4809(2016). 41 Palempalli, U. D. et al. Gambogic acid covalently modifies lkB kinase-B subunitto mediate suppression of lipopolysaccharide-induced activation of NF-kB inmacrophages. Biochem.J. 419, 401-409 (2009). ( 42 Qin, Y . e t al. Gambogic ac i d in h ibits the catalytic activity of human topoi- somerase l l alpha b y b i nding to its AT P ase dom a in. Mol. Cancer Ther. 6, 2429-2440(2007). ) .43 Kasibhatla, S. et al. A role for transferrin receptor in triggering apoptosis whentargeted with gambogic acid. Proc. Natl. Acad. Sci. USA 102, 12095-12100(2005). ( .44 Yang, J . e t al. Gambogic acid deactivates cytosolic a nd mitochondrial t h ior-edoxins b y c o valent b i nding to th e fu n ctional domain.J. Nat . Pr o d. 75 , 1108-1116(2012). ) 45 Leao, M. et al. A-mangostin and gambogic acid as potential inhibitors of thep53-MDM2 interaction revealed by a yeast approach. . Nat. Prod. 76, 774-778(2013). .46 Tian, C. et al. Multiplexed thiol reactivity profiling for target discoveryof electrophilic natural products. Cell. Chem. Biol. 24, 1416-1427(2017). ( 47 Mimnaugh, E. G . , Xu, W, Vos, M., Yuan, X. & Neckers, L. Endoplasmic reticulumvacuolization a nd v a losin-vontaining protein relocalization result fr o m simul- t aneous H sp90 inhibition b y geldanamycin an d proteasome inhi b i tion by v elcade. Mol. Cancer Res 4, 667-681(2006). ) ( .48 Al - Furoukh, N. e t a l . ClpX stimulates the mi t ochondrial unf o lded p roteinresponse ( UPRmt) i n m a mmalian cells. B i ochim. Biophys. Acta 1 853, 2580-2591 ( 2015). ) ( 49 L e e, D. M. , Kim, l . Y., Seo, M . J ., Kwon, M . R. & Choi , K. S . Nutlin - 3 e nhances thebortezomib sensitivity o f p 53-defective c a ncer cells by inducing paraptosis. Exp. Mol. M ed 49, e365 ( 2 017). ) ( 50 Clarke, H. J.,Chambers, J. E .,Liniker, E . & Marciniak, S . J. Endoplasmic reticulumstress in malignancy. Cancer Cell 2 5 , 563-57 3 (20 1 4). ) ( . 5 1. Arena, G. et al. Mitochondrial MDM2 Regulates Respiratory Complex 1 Act i vityIndependently of p53.Mol. Ce l l 69, 594-609 (20 1 8). ) ( 52. Yang, H., C hen, D . , Cu i , Q. C., Yuan,X. & Dou, Q. P. Ce l astrol, a triterpene extracted from the Chinese " Thunder of God V i ne," is a potent pro- teasome i n hibi t or a nd suppresses human prostate ca n cer growth in nude mice. C ancer R es 66,4758-4765 ( 2 006). ) ( ,53 Suh, D . H , Kim, M. K., Kim,H. S., Chung, H. H. & Song, Y. S. U nfolded proteinresponse to autopha g y as a p r o mising dru g gable target for a ntican c er ther - apy. Ann. N . Y. Acad. Sci. 1271, 20-32 (2 0 12). ) ( 54 Gupta, S. C . et a l. Upsides a nd d o wnsides of reactive ox y gen spec i es forcancer: the roles of reactive oxygen species i n tumorigenesis, p revention, and t herapy. A ntioxid. Redox S i gnal 1 6 , 1 2 95-1322 (2012). ) ( 55. Jeong, S . A. et al . Ca +i nflux-mediated dilation of the e ndoplasmic reticulum and c -FLIPL downregulation trigg e r CDDO-Me-induced apoptosis i r breast cance r cells s . . Oncotarget . 6 6 , 2 1173-21192 (2015). ) SPRINGER NATURECDDpressOfficial journal of the Cell Death Differentiation Association Official journal of the Cell Death Differentiation Association 癌症自从被发现的那一天开始就一直是困扰人类医学界的一项重大难题。人类从未终止过对癌症的研究,在癌症早期阶段,癌细胞还未扩散时,医生可以通过切除病变的部分以达到治疗癌症的方法,而一旦癌细胞扩散,医生只能通过化疗的方式来杀死癌细胞以延长换证的生命,但是在研究各种抗癌药物的效果以及作用机理时,使用荧光共聚焦显微镜对生物组织以及细胞的观察研究显得极为重要。由韩国아주大学生物医学系,生物化学与分子系,药学系,天安檀国大学医学系,韩国化学技术研究所化学数据驱动研究中心,鞍山生命科学研究所融合医学等多家机构合力研究的GA抗癌性研究获得巨大进步。藤黄酸(GA)是一种从树上的藤黄(Garcinia hanburyi)树脂中提取的类黄酮,具有抗癌活性,但其潜在的作用机理尚不清楚。在这里,我们显示GA诱导癌细胞死亡,并在体外和体内空泡化。 GA在各种癌细胞中诱导的空泡化源自内质网(ER)和线粒体的扩增,并被环己酰亚胺阻断。这些发现表明,GA通过诱导截瘫(一种空泡化相关的细胞死亡)杀死癌细胞。我们发现大线粒体的形成是由溶胀的线粒体的融合引起的,而这种融合是在ER来源的液泡融合之前。发现GA诱导的蛋白酶体抑制作用导致在治疗的癌细胞中观察到的ER扩张和ER应激,并且在线粒体膜去极化之后形成大线粒体。有趣的是,GA诱导的截瘫被各种含硫醇的抗氧化剂有效地阻断,并且这种作用与ROS的产生无关。我们观察到,GA可以与半胱氨酸硫醇反应形成迈克尔加合物,这表明GA共价修饰蛋白质亲核半胱氨酸基团的能力可能导致蛋白质错误折叠以及随后错误折叠的蛋白质在ER和线粒体内积累。研究表明藤黄酸(GA)是通过诱导截瘫(一种空泡化相关的细胞死亡)杀死癌细胞。用H&E对5μm的小鼠肿瘤细胞切片进行染色,通过使用安装在K1-Fluo上的CMOS(互补金属氧化物半导体)相机,可以观察组织切片上的图像。在分析线粒体膜电位(Δψ)时,将12孔板中培养的细胞(8×104细胞)用1μMGA或20μM(CCCP)处理, 在37°C下使用200 nM TMRM或100 nM MTR孵育30分钟。 用PBS洗涤后,在激光共聚焦扫描显微镜(K1-Fluo)下观察细胞的状态。
关闭-
1/16

-
2/16
还剩14页未读,是否继续阅读?
继续免费阅读全文产品配置单

Nanoscope Systems,Inc.为您提供《细胞中生物检测方案 》,该方案主要用于细胞中生物检测,参考标准《暂无》,《细胞中生物检测方案 》用到的仪器有 K1-Fluo ABM 激光荧光共聚焦显微镜。
我要纠错
推荐专场



相关方案
 咨询
咨询






