

heguang
第2楼2011/03/16
你说的很对,因为缓冲液中的乙腈不断挥发,然后导致分离结果不稳定。而电流上升的原因就在于乙腈挥发走了,缓冲的导电能力更强了,所以你分析的原因是正确的。
另外,由于黄酮的水溶性不好,在实验过程中不断的在毛细管里沉积,导致毛细管变脏,基线不稳,电渗流下降。这个是比较难解决的。
我的建议是对毛细管进行动态涂层,另外,缓冲里面加表面活性剂,不用乙腈,以除去乙腈挥发的影响。
如果你不是做方法开发,是想做检测的话,我建议你尝试商品化的试剂,CElixir-SDS™ 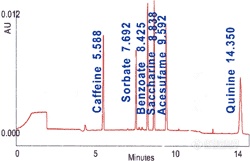
heguang
第3楼2011/03/16
Separation of Neutral Compounds by CE using CElixir-SDS™
Neutral compounds normally migrate with the Electro-Osmotic Flow (EOF) in CZE. Using the proper buffer additives, MEKC can employed and neutral compounds can be separated along with charged molecules. This pre-made 搗alidated? kit makes MEKC easy to perform, reliable and a powerful addition to any laboratory taking advantage of the power and advantages of CE.
Method Conditions:
Catalog No.: 06200-50
Supplied Buffers and Solutions were used without modification
Voltage: 25kV
Capillary: MicroSolvCE™ Bare Fused Silica, 75祄 x 60cm
Injection: 5 sec, 0.5psi followed by post injection Water Plug
Run Buffer: CElixir-SDS Accelerator™ 50-50 Borate pH 9.2
Detection: UV, 200nm
Polarity: Normal
Sample Preparation:
A standard Test Mix of Caffeine, Sorbate, Benzoate, Saccharine, Acesulfame and Quinine was used.
Discussion and Rationale:
The CElixir-SDS™ kits use the patented dynamic coating technology of CElixir™ which makes getting results easier and more reliable. Precision through stabilization of the EOF and control of the SDS allows these kits to be used by beginners as well as experienced experts. No buffer preparation and therefore less variation in critical CE parameters also makes these kits the choice of routine as well as research laboratories.
babylon
第4楼2011/03/16
我觉得楼上的能否提供点可行的方案而不是试剂;事实上我是想尝试用CD来改善黄铜的分离,因为别人都是用的SDS,而我的师兄用CD分了几个东西还比较成功,而我这里面的9个很类似的黄铜在我的BARAX-CD系统里加如乙腈后,分离度是大大改善了。现在就是峰漂移的问题。我现在已经找了一个原因出来,乙腈的挥发是导致峰顺序改变的主要原因。经常换BUFFER可以解决,另外我可以用稍微低挥发的异丙醇来替代乙腈
另外就是吸附的问题。我的黄铜的水溶性很好,本身我就是用流动相来溶解的。因此我想这个问题应该不是主要的问题吧。此外,在样品间加NAOH冲洗是否可以改善??
babylon
第5楼2011/03/16
我觉得楼上的能否提供点可行的方案而不是试剂;事实上我是想尝试用CD来改善黄铜的分离,因为别人都是用的SDS,而我的师兄用CD分了几个东西还比较成功,而我这里面的9个很类似的黄铜在我的BARAX-CD系统里加如乙腈后,分离度是大大改善了。现在就是峰漂移的问题。我现在已经找了一个原因出来,乙腈的挥发是导致峰顺序改变的主要原因。经常换BUFFER可以解决,另外我可以用稍微低挥发的异丙醇来替代乙腈
另外就是吸附的问题。我的黄铜的水溶性很好,本身我就是用流动相来溶解的。因此我想这个问题应该不是主要的问题吧。此外,在样品间加NAOH冲洗是否可以改善??
heguang
第6楼2011/03/16
看来你是打算做方法学研究。
你的峰不稳定是怎么样的变化?是出峰时间变长还是变短?
babylon
第7楼2011/03/16
没有错,我是打算做方法的研究的。同时分离八种物质;事实上我的 体系是可以实现分离的。无论是应用在对照品还是真样品中效果都非常的好。问题就在于保留时间的重现性上。
如果每次都是一根一模一样的新柱子的话,都是一模一样的新buffer的话,我的实验基本是可以重现的。问题在于重复进样以后出现了以下问题:
1,保留时间靠后的峰漂移很厉害。比如最后一个峰本来该是32分钟出来,有可能进了10针以后就变成了27分钟。
2,峰顺序的变化。这个也出现在靠后出来的几个峰。明显的往前移动。
3.奇怪的是,还出现相邻的两个峰集体移动。我认为这个是由于EOF的变化引起的,而且可能和有机溶剂的挥发有一定的关系。
4。当进了很多REAL sample以后,原本非常sharp的峰明显的底部变宽,有点拖尾的样子。就是类似于HPLC里面柱效下降的样子。因为这样导致了分离度的下降,导致相邻的峰峰底部重叠。由于靠后的峰又往前移动,导致和前面的峰又有分不开。
babylon
第8楼2011/03/16
我有分析我的重复性不好的原因:
1。刚才说的部分是由于有机溶剂的挥发导致的。这个我可以证实。因为用平行放置的BUFFER和新配的BUFFER是效果不一样的。新配的BUFFER可以将保留时间拉回最初的时间段。但是新配BUFFER无法解决柱效降低的问题。应该这样讲,相对于平行放置的BUFFER(溶剂挥发掉一些),新配的含有有机溶剂的BUFFER 柱效要差一些。但是如果不加有机溶剂的话,整个有效保留时间无法解决待测峰和杂质峰重叠的问题。尤其是保留时间靠前的物质。
2。样品污染柱子的问题。事实上我的样品是用BUFFER 溶解的。所以应该不会有沉淀析出的问题吧?在这之前我曾经用甲醇溶解过样品,但是出现堵柱子的情况,我就已经换成了BUFFER溶解了。但是部分的竞争性吸附我不排除,在样品与样品间我有加0.1M的NAOH冲洗。
3。缓冲溶液浓度太高的问题,导致焦耳热,导致住效下降。原本这个问题我是没有想到的,一个网友提到这个。他说我的BARAX的浓度有点高50mM。那么毛细管在长时间运行的时候就会发生柱效降低的情况。这个我不太清楚其中的道理,因为我的是BECKMAN的仪器,应该说温控效果还是可以的。
4。样品前处理的问题。我的样品只是简单的用80%甲醇提取,然后挥干,用BUFFER溶解。里面可能物质比较多,容易脏。这个和液相色谱类似。于是有人提醒我让我做一下前处理,只要黄酮不要别的。事实上我发现出来的峰基本上不是黄酮就是多酚。因为光谱里面可以看得出来的。再加上电泳的选择性。
我只想到这么多了。有朋友给点建议吗?我这个系统应该如何改?
其实CD的分离效果和效率是不容置疑的,相比SDS的话峰型也好。我想我现在要解决的是稳定性的问题。希望各位给我一点建议。
heguang
第10楼2011/03/17
像是乙腈挥发及样品沉淀产生的影响,乙腈挥发导致EOF上升,出峰变快,分离效率下降。同时buffer中的乙腈降低,样品溶解度变差,容易在毛细管上有吸附,产生峰的拖尾和变宽。
建议:
1、样品与样品之间加入乙腈冲洗过程,以洗掉样品在毛细管里的沉积;
2、频繁更换新的缓冲液(buffer要密封保存)。

































