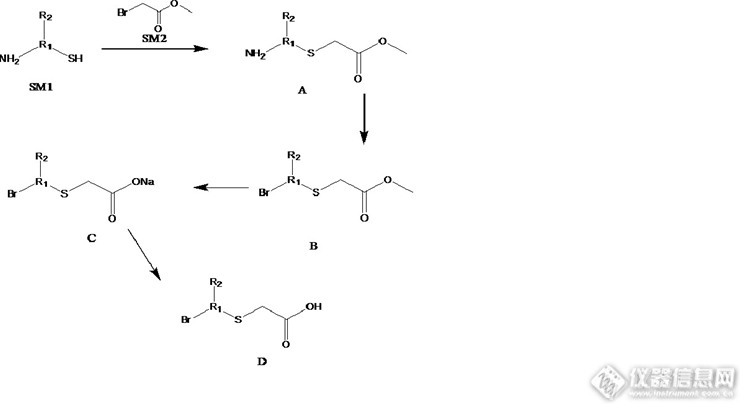
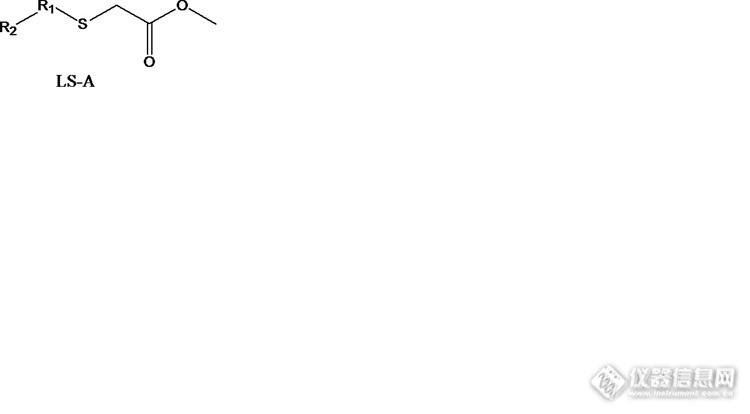
化学药检测
浪淘沙隐
第1楼2015/08/31
图片没设置好,各位将就看下。
小鱼625
第2楼2015/08/31
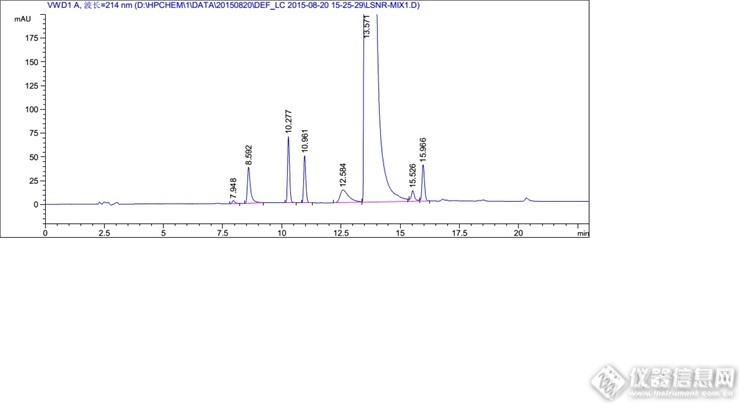
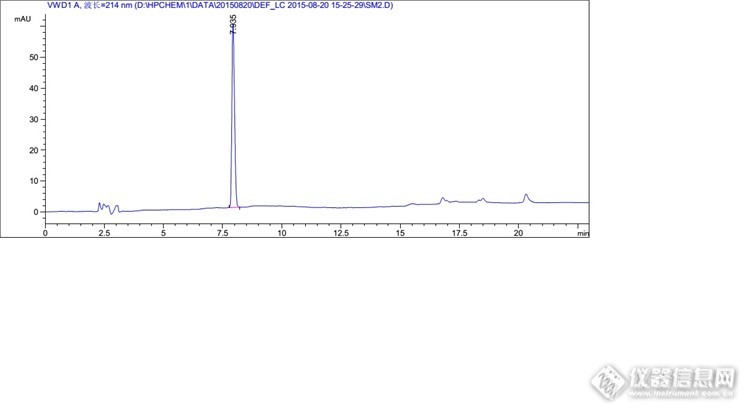
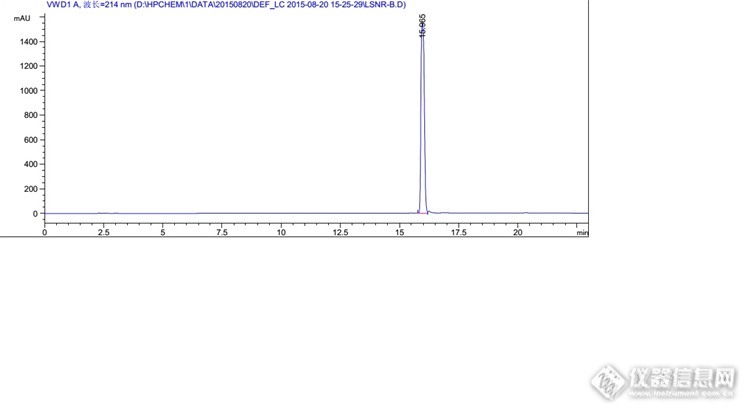
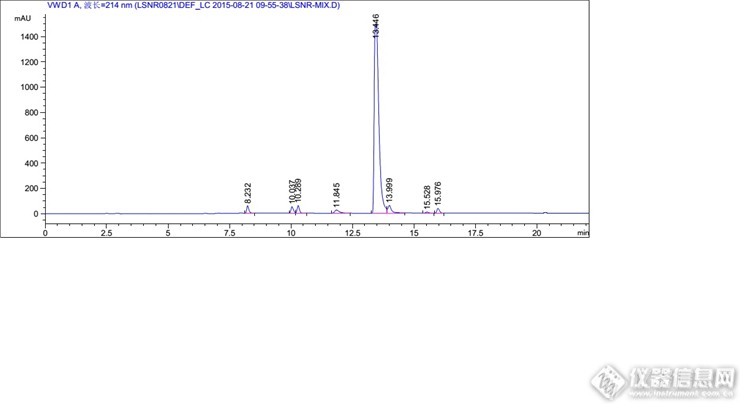
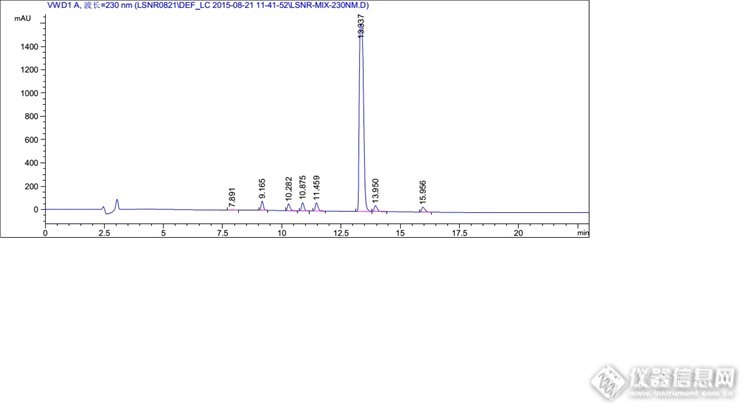
分析思路很好,楼主开发研发能力很强啊。 那个全景图,有一个峰特别高,其他的都相对很低,可以调试下浓度,让峰型更美观些否?
第3楼2015/09/01
不敢说强,论坛估计有很多比我强的。我这个混合溶液的杂质是按1%的浓度配制的,目的是考察出峰情况,保证能清晰的辨认出峰情况,很多系统适用性也是按照这个浓度来考察的,所以不好调浓度。其实这里我上文也提到了,1mg/ml的溶液,主成分是过载的,根据实验,最后把主成分浓度降低到了0.3mg/ml,那么杂质浓度相应的也会降低到约为以前的1/3。
wyong5053132
第4楼2015/09/01
很好的学习资料!
第5楼2015/09/06
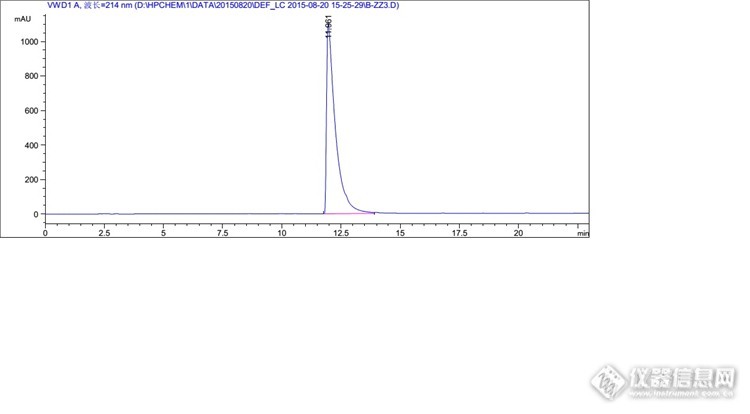
其实这个例子里,我是走了一些弯路的。比如进第一针,峰形不好,那么这时候是没必要进行单针定位的,根据主峰的峰形和分子结构就可以判断,直接换流动相调整好峰形再定位就可以了;另外0.1%磷酸体系是可以不试验的,因为0.1%磷酸pH大概是2.4,与pH3.0的流动相相比变化不大(当然试验一下也是可以的,毕竟这个方法波长比较低,如果能用磷酸,那么基线漂移就会好点)。
trees83
第6楼2015/09/21
受教了!很感谢!
Insm_b97c118a
第7楼2022/01/01
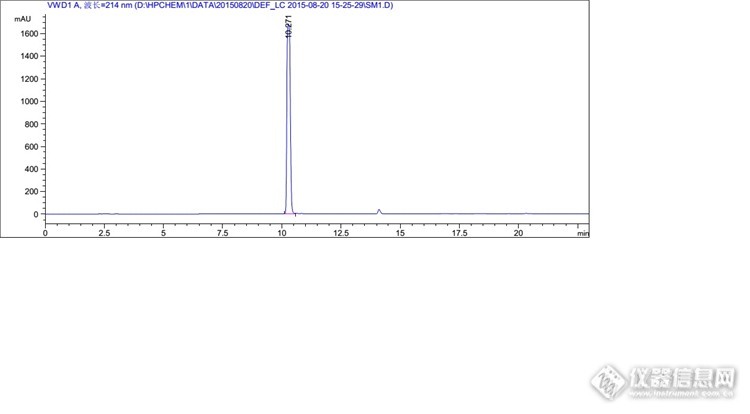
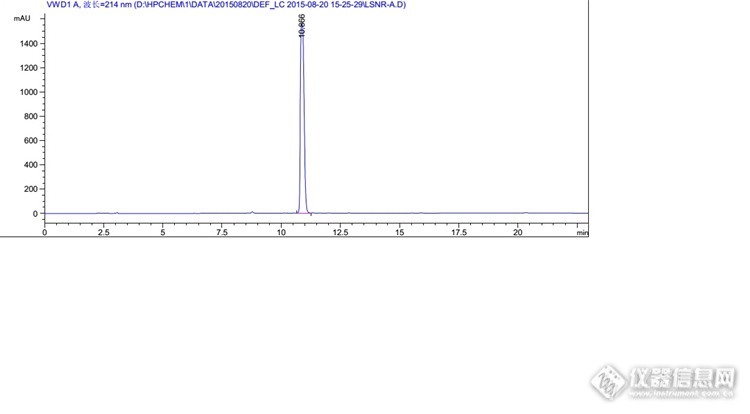
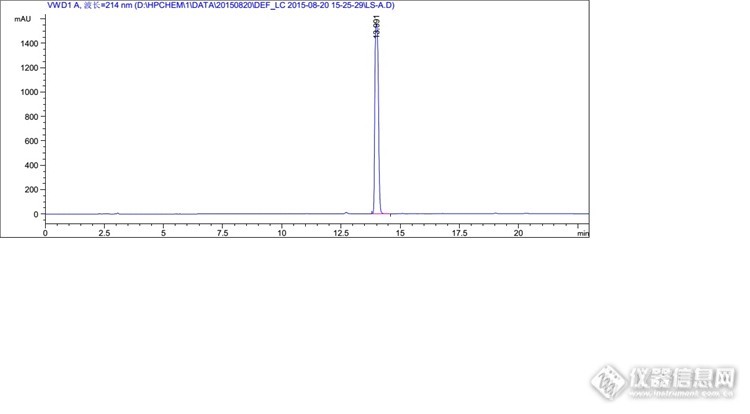
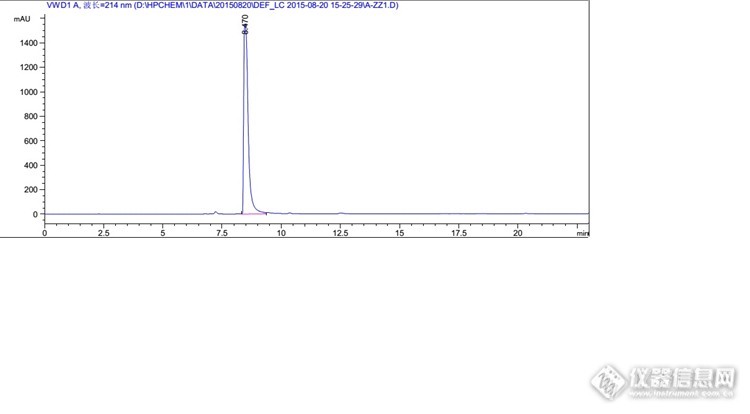
你好,我想问一下你这里面说的峰过载是怎么判断的
第8楼2022/01/26
1、峰高超过1000就有可能过载;2、峰平头或不够尖锐,基本上就是过载了;3、降低浓度或减少进样量后,峰面积不呈线性了,差不多就是之前过载了。
品牌合作伙伴
执行举报