


大家晚上好,我是月旭科技的刘香艳,很荣幸今天晚上能够在这里跟大家一起讨论气相柱的相关问题。
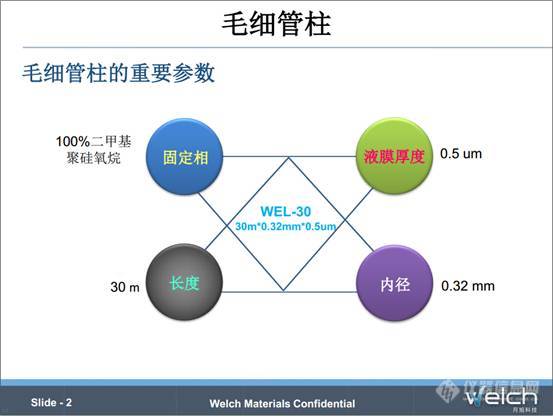
我们知道在日常的分析检测中,GC是一种很重要的分析方法。气相的应用也极为广泛,从制药行业、食品检测,到环境保护、石油化工,面对这些应用,选择气相柱是一项非常重要而且复杂的任务。既然要选择气相柱,我们就先从四个重要的色谱柱参数说起,看看他们是如何影响气相柱的性能的。我们看一下这张PPT上的例子,WEL-30 30m×0.32mm×0.5um,这就是毛细管色谱柱的完整描述,包括固定相(WEL-30)-----100%二甲基聚硅氧烷,长度-----30m,内径-----0.32mm,液膜厚度-----0.5μm这四个参数。

选择毛细管柱的时候,最重要的是选择一个合适的固定相。固定相从两个方面影响毛细管柱的性能,一方面是固定相的选择性,选择性可以认为是固定相区分两种待分离物质的物理、化学性能差异的能力,取决于固定相和待分离组分的作用力区别;另一方面就是固定相的极性,取决于固定相自身的结构。
我们先看一下固定相的选择性,不仅仅和固定相的结构有关,和待分离的组分性质也有关,取决于固定相和待分离组分的相互作用力,作用力不同,则可以分离。对我们常用的毛细管固定相(聚硅氧烷类和聚乙二醇类)来说,和待分离物质存在三种主要相互作用:色散力、偶极力以及氢键作用力。

固定相的选择性,也就是固定相如何和组分发生相互作用。我们从毛细管柱固定相的结构来判断各种作用力的强弱,我们知道,毛细管柱主要的两类固定相:聚硅氧烷和聚乙二醇。聚硅氧烷包括100%二甲基聚硅氧烷(绿色结构式),苯基取代聚硅氧烷(无色结构式)以及氰丙基苯基取代聚硅氧烷(黄色结构式)。聚乙二醇是极性的固定相(黄蓝色结构式)。固定相的特征官能团的相互作用总结见PPT中表格,色散力是所有聚硅氧烷和聚乙二醇固定相都存在的相互作用力,而偶极力和氢键作用力只存在于氰丙基取代聚硅氧烷和聚乙二醇类固定相中。
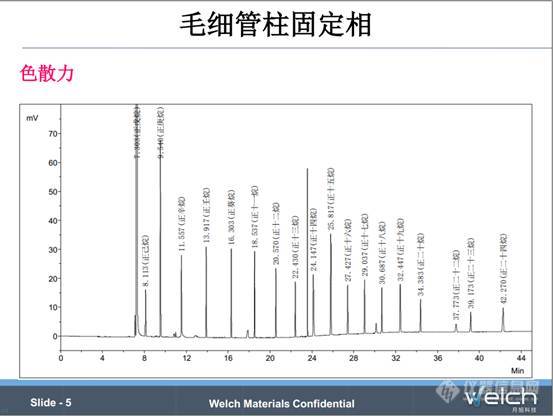
色散力是聚硅氧烷和聚乙二醇都具有的主要相互作用力,色散力可以简单认为挥发性,也就是可以简单用沸点来衡量。对于具有简单结构的同系物,用色散力简化方法判断物质的出峰顺序比较有效,我们看PPT中色谱图,这是用100%二甲基聚硅氧烷的非极性柱分离C5-C24的烷烃同系物,组分越容易挥发,越早流出色谱柱 。但是面对具有不同结构的组分,单纯用挥发性判断出峰变得没有规律。

用色散力简化方法判断具有不同结构的物质的出峰往往会出现失误。PPT中谱图是用100%二甲基聚硅氧烷的非极性柱分离甲苯、己醇、苯酚、癸烷、萘、正十二烷六种组分,苯酚(沸点182℃)出峰早于癸烷(沸点174℃),萘(沸点219℃)出峰早于正十二烷(沸点216℃),这和色散力简化方法推断的出峰顺序是矛盾的。
总结一下,色散力只能判断具有简单结构的同系物的出峰,对于沸点相差30℃的组分,选用大部分固定相通常都可实现分离。
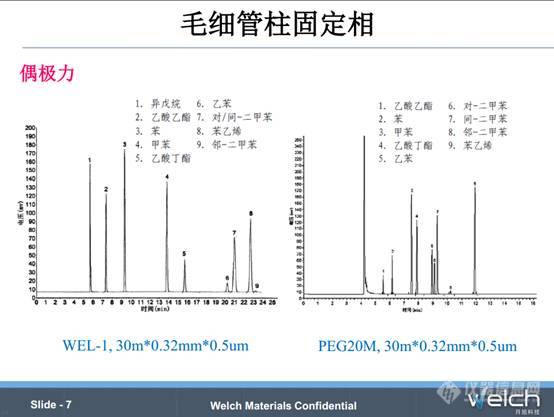
只有氰丙基取代聚硅氧烷和聚乙二醇类固定相具有偶极力相互作用,使用这类固定相的色谱柱时,偶极矩不同的溶质可以实现更好的分离。PPT中的应用谱图,对二甲苯和间二甲苯分子量和沸点几乎没有区别,只有甲基的相对位置不同导致的偶极相互作用不同,只有具有强偶极作用的固定相才可以实现分离。用100%二甲基聚硅氧烷的固定相色谱柱分离时,对二甲苯和间二甲苯的分离度很小,换用聚乙二醇的色谱柱,两种组分的分离度有很大改善。
如果两种组分的偶极作用区别较小,选用气相柱时,应该使用偶极作用更大的固定相,预测组分的分离程度的变化比较困难,通常需要做实验确定。

和偶极力相互作用类似,只有氰丙基取代聚硅氧烷和聚乙二醇类固定相才可以充分利用偶极力相互作用。使用这类固定相时,具有不同氢键作用力的组分能够实现更好的分离。PPT中表格列出了能够形成氢键的组分及氢键的作用力强弱。
PPT中谱图,分别用非极性固定相(100%二甲基聚硅氧烷)和极性固定相(聚乙二醇20M)分离甲苯、己醇、苯酚、癸烷、萘、正十二烷六种组分,由于甲基取代基不具有氢键力相互作用,己醇、苯酚在极性柱上由于和固定相的偶极、氢键力相互作用的共同影响,出峰时间更晚。

毛细管柱依据极性的不同,分为非极性、弱极性、中等极性和极性四种。固定相的极性取决于取代基的极性和含量。毛细管柱的极性,是影响分离的重要因素之一。通常来说,极性固定相对极性组分的保留更强,弱极性固定相对于弱极性组分的保留更强。
另外毛细管的极性也影响毛细管柱的惰性,一般情况,固定相的非极性越强,色谱柱的寿命、温度上限越高。这里要提醒一下,对于极性固定相,容易出现氧损坏的问题,导致组分峰形拖尾,柱效降低。所以需要注意一些事项:载气的纯度要达到99.999%以上,不能用气体发生器作为气源;最好加上脱氧管;柱温升高之前,一定要先通载气20min左右;柱温不能超出色谱柱的温度上限。
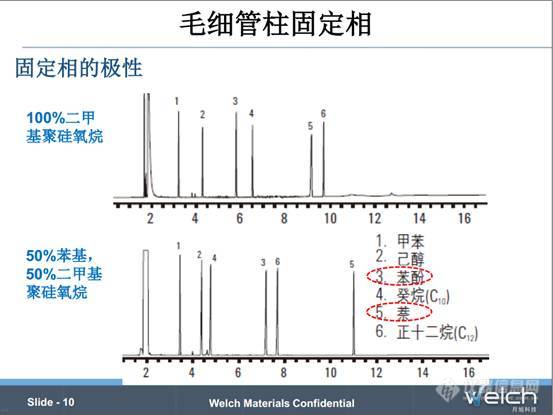
PPT中两张图谱,,分别是用100%二甲基聚硅氧烷的色谱柱和50%苯基-50%二甲基聚硅氧烷的色谱柱,后者的极性强于前者。所以对于50%苯基-50%二甲基聚硅氧烷的色谱柱,苯酚(极性)的保留相对于癸烷(非极性)的保留会增强。

从毛细管固定相的选择性和极性出发,对固定相的选择做一个总结:1.主要参考色谱柱制造商的应用实例、药典以及国标。2.从成本方面考虑,能够满足分离要求的前提下,尽量选择寿命更长的非极性柱。3.固定相对于极性相近的组分通常有更强的保留,根据固定相对不同组分保留能力的差别,实现分离。4.如果新做一个项目,没有资料可以参考,可以先从非极性柱或弱极性柱开始尝试。

接下来我们看一下色谱柱长度对分离度和保留时间的影响。从PPT中三个关系式看到,分离度R和理论塔板数(N)的平方根成正比,柱效(N)与柱长(L)成正比,所以分离度是色谱柱长度的平方根函数。如果将柱长增加一倍,理论上分离度会提高1.41倍,实际分析中,分离度提高1.2-1.3倍。
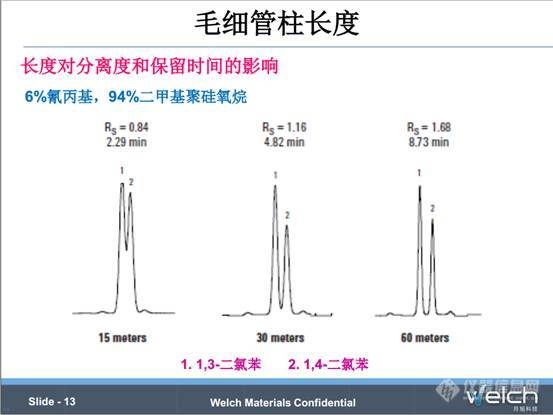
这是一个长度对分离度和保留时间影响的一个例子。分别用15m,30m和60m长的色谱柱分离1,3-二氯苯和1,4-二氯苯。随着长度的成倍增加,保留时间也在成倍增加,而分离度增加的倍数只有 倍。
倍。
另外,长度加倍,成本也加倍;色谱柱的流失也是随着柱长的增加而增加;所以想要通过增加柱长增加柱效时,需要综合考虑这些因素。

长度的选择总结:1.用25-30m长的色谱柱能完成大部分常规分析;2.如果待测体系中含有的组分比较少,可以选用10-15m长的色谱柱;3.如果分析样品中含有的成分比较复杂,或者含有不容易分离的组分,可以选用50-60m长的色谱柱。
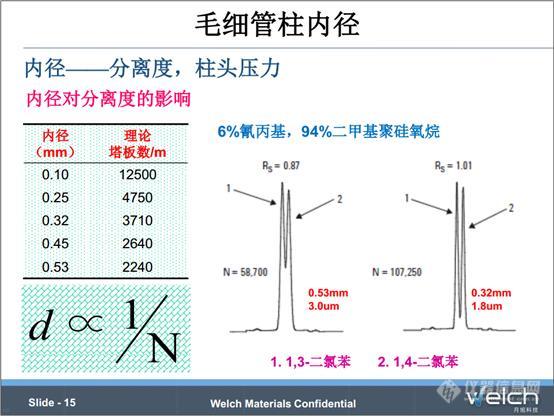
我们看下色谱柱内径的影响。PPT中表格数据(数据来源于安捷伦文献)显示了理论塔板数随色谱柱内径的变化规律,直径越小,柱效越高,柱效和色谱柱内径呈反比关系。理论上,将色谱柱的内径减小一倍,柱效就增加一倍,即分离度增加0.41倍。
我们看下PPT中0.53mm和0.32mm色谱柱分离1,3-二氯苯、1,4-二氯苯的对比谱图,用0.32mm色谱柱,峰型更窄。不过这个例子里边还有膜厚的影响,我们通过这个例子了解下内径对柱效的影响规律。

内径对柱头压的影响可以从PPT中公式推荐,在其他条件相同的前提下,内径是0.25mm的色谱柱的柱头压是内径0.53mm色谱柱的4.5倍。
内径不同,分析样品的时候所采用的载气流速大小也不同。表格中列出了推荐的载气流速,可以参考一下。如果载气流速太小,柱温升高时可能对色谱柱有损害,也有可能引起仪器报警;如果载气流速太大,会影响某些组分的分离,所以载气流速选择要合适。

毛细管柱内径选择总结:(1)比较常用的规格有0.25mm和0.32mm,与0.32mm内径的色谱柱相比,0.25mm内径的色谱柱具有更高的柱效,但是样品容量稍小;(2)0.45mm内径的色谱柱比较适合用于顶空进样、阀进样等;(3)0.53mm内径的色谱柱,样品容量比较大,比填充柱有更高的柱效,正在越来越多的替代填充柱,特别适合于微量组分的分析,提高微量组分的分析灵敏度。
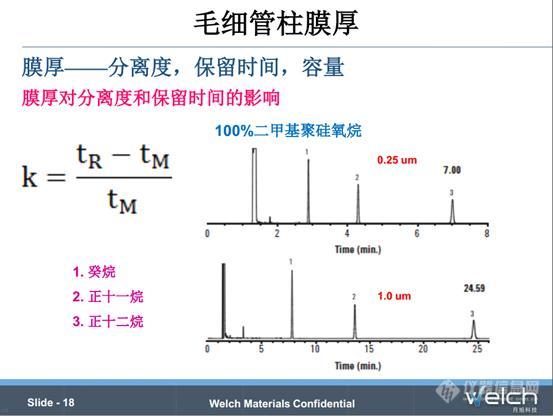
膜厚主要从分离度、保留、容量、惰性、流失等影响色谱柱的性能。我们先来了解一个概念,保留因子(k)是组分在固定相和流动相中保留时间的比值,k是衡量某组分在固定相中的保留能力,k越大,组分在固定相的保留能力越强。
PPT中谱图是两个不同膜厚的柱子分离癸烷、正十一烷、正十二烷的情况,膜厚的色谱柱对三种组分的保留更强。通常来说,易挥发的组分在厚膜的色谱柱中才能得到充分保留,液膜较厚的色谱柱通常用于易挥发物质的分析,反过来,薄膜的色谱柱常用于高沸点组分的分析。所以增加膜厚可以改善较早流出组分的分离度,但是可能会减小较晚流出组分的分离度。
对于k>10的组分,说明该物质在固定相中的保留能力比较强,如果再增加膜厚,可能对改善分离度作用不大;对于k<5的组分,增加膜厚通常可以增加其分离度;对于k在5-10之间的组分,增加膜厚可能稍微改善其分离度。
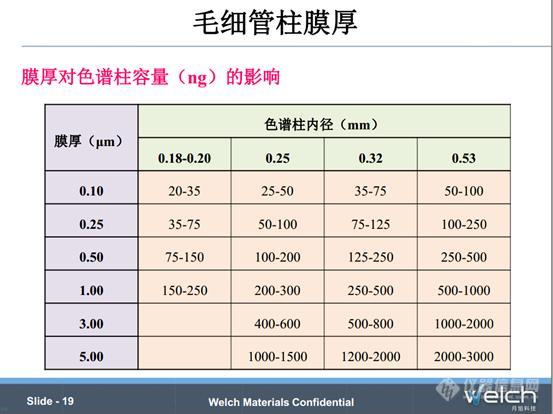
膜厚和内径对色谱柱的容量的影响见PPT中表格,膜厚越大,色谱柱的溶质容量越高。膜厚较厚的色谱柱通常可以减少峰展宽,改善活性化合物的峰拖尾。
从柱流失来考虑,膜厚增加,柱流失也会增加,较晚流出的峰可能在色谱柱流失较高的地方,厚膜色谱柱的温度上限会降低。

膜厚选择总结:对于大多数分析,色谱柱内径小,膜厚相对较小;厚膜适合于分析挥发性组分,薄膜适合分析高沸点组分。
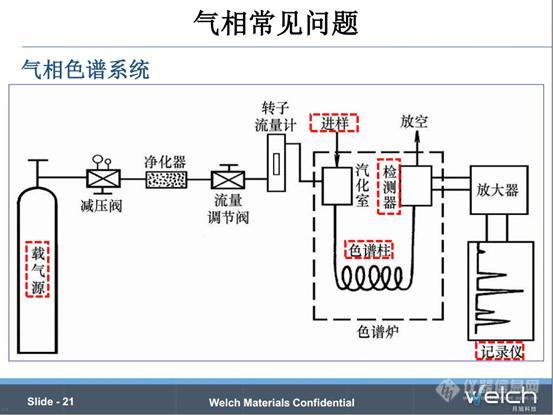
上述内容是色谱柱的4个重要参数(固定相、长度、内径以及膜厚)对色谱柱分离度、保留等性能的影响,下边我们看一下气相中常见问题。
气相的整个系统,包括五大组成部分。1.气体,包括载气源和检测器气体;2.进样口,也就是样品引入和汽化的系统;3.色谱柱,也是分离的核心。4.检测器,包括氢火焰离子化检测器(FID)、热导检测器(TCD)、电子俘获检测器(ECD)和火焰光度检测器(FPD)等;5.数据处理系统。
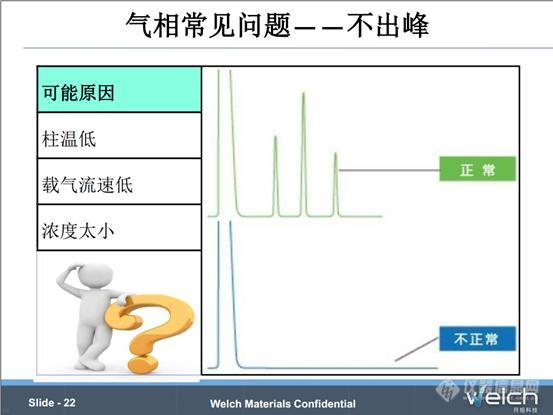
气相中如果只出现溶剂峰,可能的原因有进样针问题、载气泄露、组分和溶剂没有分离、样品浓度太低或者柱温载气流速不合适。下边我们看两个例子。

这个是2015版《中国药典》二部中薄荷脑的含量测定,用的是WM-PEG20M 30m×0.32mm×0.5μm,第一个谱图用的柱温是120℃,在我们采集的15min内薄荷脑没有出峰,我们发现在在进下针样品的时候出现一个额外峰,而薄荷脑的沸点是212℃,所以判断是薄荷脑出峰时间晚于15min。第二个谱图我们采用了PPT中的程序升温,薄荷脑的出峰时间提前到7.1min。

这个也是薄荷脑的含量测定谱图,这次测试我们没有改变柱温(120℃),而是将色谱柱柱前压提高到0.1MPa,与提高柱温类似的效果,薄荷脑的出峰时间提前到4.9min。
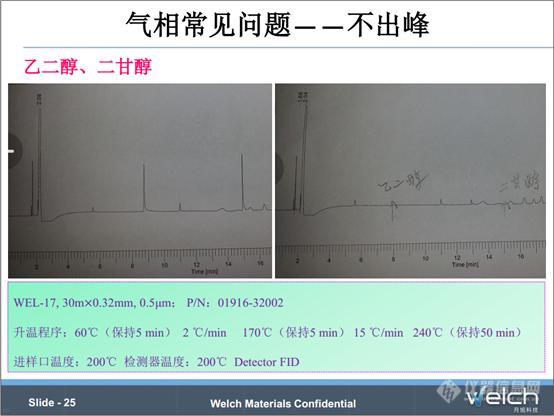
这个是2015版《中国药典》四部中聚山梨酯80项下乙二醇与二甘醇的测定,第一张谱图乙二醇与二甘醇的浓度都是0.4 mg/ml,而第二张谱图乙二醇与二甘醇的浓度都是0.04 mg/ml,在同样的色谱条件下,浓度高的出峰,浓度低的目标峰没有出来。面对由于浓度低导致不出峰的问题,我们可以采取增大进样量、减小分流比等方法来解决,目的就是为了增大色谱柱中的目标组分的含量。
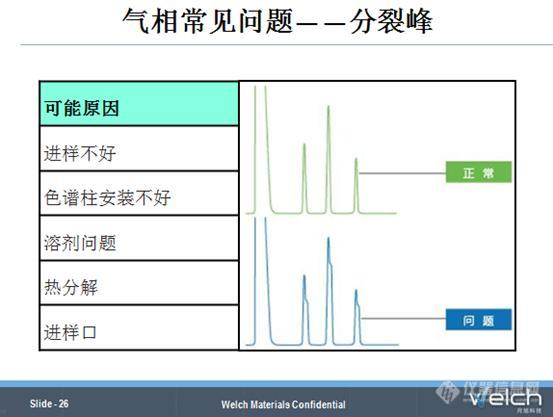
分裂峰也是气相中的一个常见问题,可能的原因有:手动进样动作不稳定,进样时要求连续快速,使用自动机样器能很好的解决这个问题;色谱柱在进样口的安装位置不合适,色谱柱高出石墨垫圈4-6 mm比较合适,此时需要重新安装色谱柱;样品萃取过程和稀释过程使用的溶剂不一样,尽可能使用同一种溶剂;不分流进样和柱上进样,容易出现样品聚焦不充分,导致分裂峰,打开分流或者调整汽化温度;进样口出现污染,对组分产生吸附,出现“二次进样”,导致分裂峰,需要进行进样口维护,如更换进样口隔垫、衬管以及分流镀金平板;进样口温度太高,导致某些组分发生热分解,需要调整进样口温度或者调整分析方法。
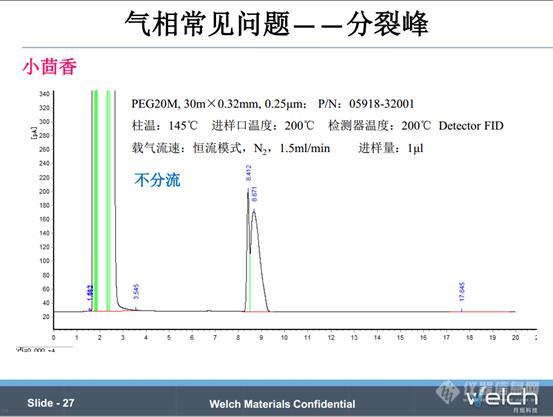
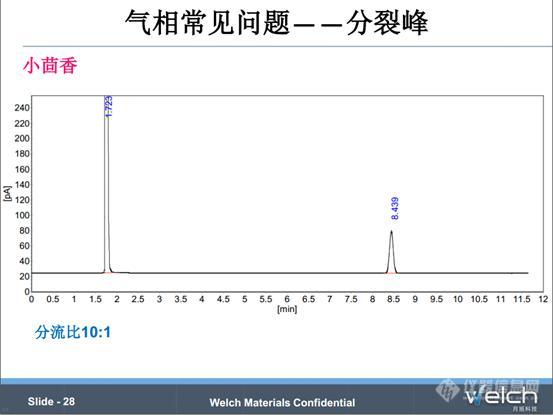
这个是2015版《中国药典》二部小茴香项下的含量测定,按照药典条件,不分流进样,结果出现分裂峰。通过进溶剂空白排除了进样针污染、进样衬管污染的原因。第二张谱图,我们将分流比设置为10:1,分裂峰问题解决,推断分裂峰应该是由于样品没有充分聚焦造成的。经验表明,不分流进样容易出现峰拖尾,峰展宽等色谱柱过载问题,所以实际操作中我们最好开分流阀,改善峰型,同时延长色谱柱的使用寿命。

气相中峰面积变小,也就意味着灵敏度的损失,通常有两种情况:所有组分的峰面积都变小和某些组分的峰面积变小。所有组分的峰面积变小的原因有,需要检查进样针无堵塞和泄露、检查色谱柱连接进样口检测器端口没有漏气、检查检测器流量电压及其他问题;某些组分的峰面积减小,要观察下这些组分是否有相似的挥发性和官能团,如果这些组分是易挥发组分,要检查下样品瓶或进样针是否泄漏、样品制备过程中是否有挥发,如果这些组分是高沸点组分,可能是因为样品没有充分挥发,此时需要提高进样口温度,玻璃衬管中装入玻璃棉。

这个是四羟乙基乙二胺,沸点280℃左右,是一个高沸点组分。当时一个客户选用了SE-30 30m×0.53mm×7.0μm的色谱柱,出现的问题是重现性很差,第一次进样,四羟乙基乙二胺的含量是74.59%,而第二次进样,该组分的含量减低到58.06%,而第三次进样,该组分就基本不出峰。我们知道,膜厚的色谱柱对高沸点的组分保留能力比较强,所以我们需要换成膜薄的色谱柱减小四羟乙基乙二胺的保留。
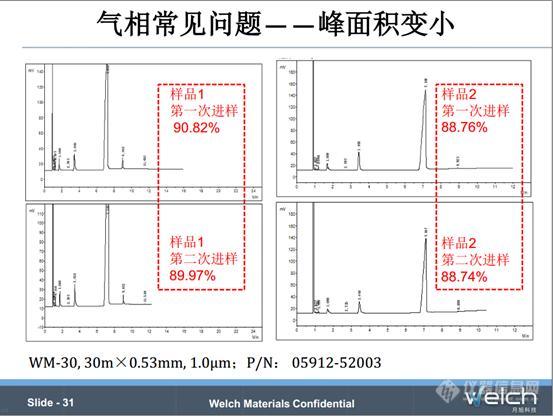
我们做了一次样品测试,换成WM-30 30m×0.53mm×1.0μm色谱柱。我们看到样品1和样品2的结果,重现性比较高。
高沸点组分,容易残留在色谱柱中,形成一层“新”的固定相,该组分在固定相和流动相中的分配比例会发生一些变化,影响重现性。建议定期进行进样口维护和色谱柱的老化,有助于延长色谱柱的使用寿命,也提高分析的准确性。
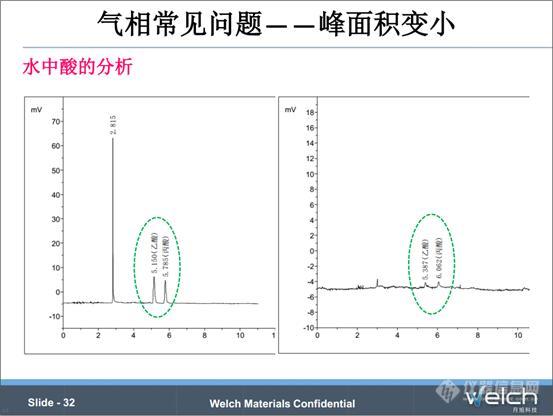
我们再来看一个例子,水中酸的分析,FID检测器,直接进样。经过几次进样,乙酸和丙酸的峰变小很多,重现性很差。水做溶剂时,一方面会对FID的火焰有影响,另一方法,可能会残留在色谱柱中,对后一针样品有吸附作用,造成峰的重现性差。
针对水做溶剂的样品分析,我们建议用顶空进样,因为水对有些色谱柱有损害,但是损害程度我们很难预测,也可以选用其他耐水的色谱柱,如WM-INOWAX、PLOT色谱柱以及GDX系列填充柱。为了提高水中组分分析的重现性,我们建议在原来柱温的基础上加一个高温段,建议以10℃/min的速率升高到200℃保持15min左右,这个主要是为了每次进样以后将柱子里面的水都除干净,不影响下一次进样。
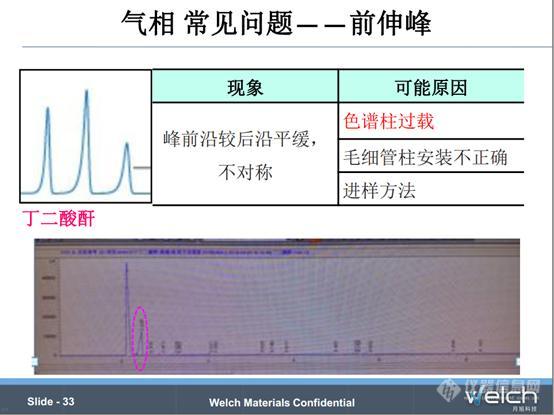
气相中不对称峰包括前伸峰和拖尾峰,前伸峰是指峰前沿较后沿平缓,拖尾峰是指后沿较前沿平缓。造成峰前伸的主要原因是色谱柱过载,这时我们需要降低进样到色谱柱中分析组分的量,比如减小进样体积、提高分流比、稀释样品;另外手动进样大体积样品、毛细管柱安装不合适也会造成前伸峰的问题。
我们来看一个例子,有老师做丁二酸酐的检测,丁二酸酐峰前沿,对比我们测丁二酸酐样品的条件,通过增大分流比、降低载气流速解决了问题。
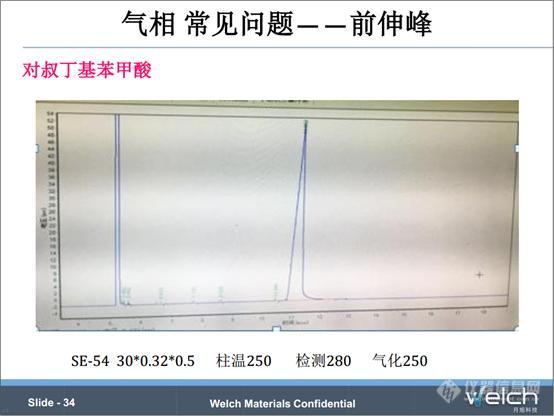
对叔丁基苯甲酸的气相检测,这个组分的沸点是280℃。从色谱条件来看,进样口温度是250℃,将其提高到280℃,峰型得到改善。这个物质更适合用液相分析。

峰拖尾,也就是说峰后沿较前沿平缓,是不对称峰。造成峰拖尾的因素有柱污染或损坏,此时需要将色谱柱的柱头切掉10-20cm;内衬管或玻璃棉污染,对样品组分产生不必要的吸附;色谱柱安装不合适造成进样口或检测器的死体积较大;两个化合物共洗脱,比如含量相对小的成分,峰被包在相邻主峰之中;进样量过大,可能会出现样品聚焦不充分的情况。

另一个例子是溴甲烷,排除了色谱柱污染、内衬管、色谱柱安装问题,建议老师将分流比调大到80:1-100:1,峰对称得到改善。
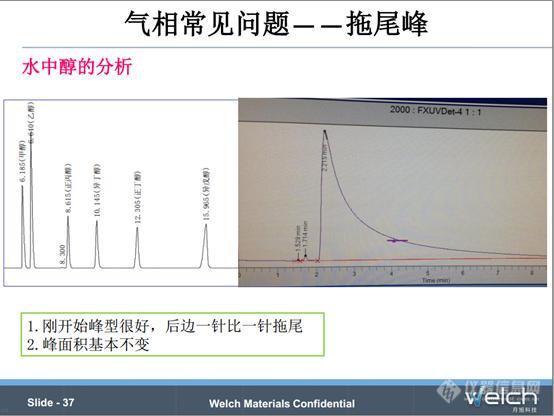
水中醇的分析,使用PEG20M色谱柱,直接进样,刚开始峰比较对称,后边峰拖尾越来越严重,峰面积基本不变,我们简单称为水的溶剂效应。这个跟水中酸分析一样,最好用顶空进样;如果没有顶空进样装置,建议在原来柱温的基础上加一个高温段,建议以10℃/min的速率升高到200℃保持15min左右,这个主要是为了每次进样以后将柱子里面的水都除干净,不影响下一次进样。
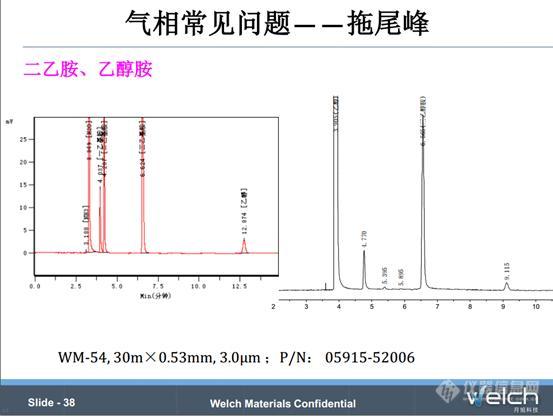
二乙胺、乙醇胺的分析,这类组分属于活性组分。我们前面讲过,膜厚的色谱柱具有更好的惰性,固定相在管壁上覆盖更完全,从而避免了管壁上的活性位点对活性化合物的吸附,所以针对二乙胺、乙醇胺等活性组分,最好选择膜厚的色谱柱,如WM-54 30m×0.53mm×3.0μm,改善峰拖尾现象。

药典或者国标中往往规定分离度、柱效要求,分离度达不到要求或降低,都有哪些可能的原因呢?(1)色谱柱参数(固定相、长度等)、色谱条件(载气流速、柱温)等选择不合适;(2)色谱柱污染或固定相流失造成分离度下降;(3)样品浓度变大,导致分离度下降。

溴乙酸乙酯、溴乙酸丁酯、二甲亚砜(DMSO)的气相分离。当时老师用DB-FFAP 30m×0.32mm×0.5μm气相柱,第一张谱图看到溴乙酸乙酯和DMSO的分离情况,第二张谱图看到溴乙酸乙酯和溴乙酸丁酯的分离情况,而老师希望DMSO作为溶剂,分析溴乙酸一直和溴乙酸丁酯,我们看到在同样的色谱条件,溴乙酸丁酯和DMSO的出峰时间很接近,都在10.5min左右。如果DMSO作为溶剂,浓度变大,峰展宽,和溴乙酸丁酯的出峰时间更接近。为了达到老师的要求,我们尝试更长的色谱柱,比如50-60m。
当时考虑到极性柱相对非极性、中等极性色谱柱来说,温度上限较低,更容易出现氧损坏,我们换用WEL-1701这个中等极性的柱子,实验结果表明,用DMSO作溶剂,溴乙酸乙酯和溴乙酸丁酯可以得到很好的分离。
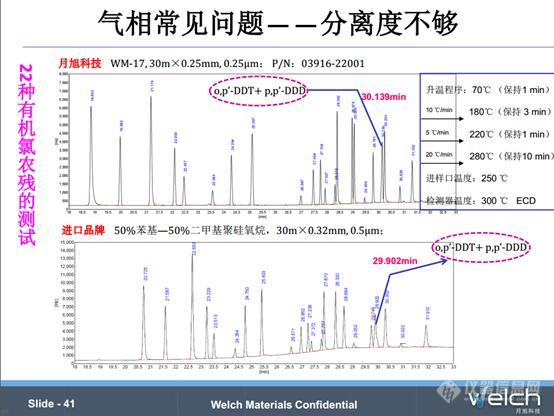
2015版《中国药典》四部中22种有机氯的分析,第一张谱图是我们月旭科技WM-17色谱柱测试结果,第二张谱图是著名的进口色谱柱的测试结果,对比谱图发现月旭科技色谱柱和进口色谱柱的分离性能相当,而且都存在一个问题,有两种物质分离不开,即o,p’-DDT和p,p’-DDD。
我们可以得到这样一个信息,o,p’-DDT和p,p’-DDD的出峰温度在280℃,而o,p’-DDT的沸点在260℃,推断有可能是出峰温度太高导致两种物质的分离度不够。
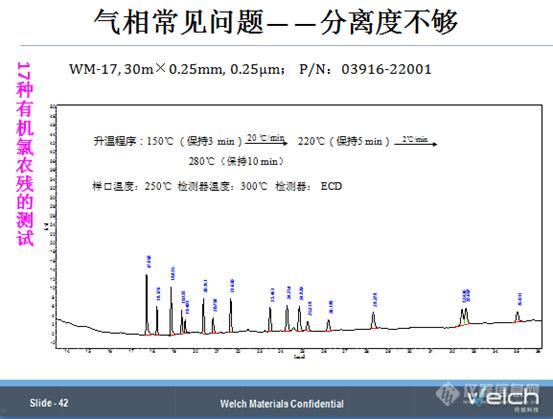
接下来,应一个老师的要求,做了17种有机氯农残的分析,我们调整了一下升温程序,使得o,p’-DDT和p,p’-DDD的出峰温度在260℃左右,两者的分离度提高到1.0左右。
当然我们用WM-1701这个固定相的色谱柱,o,p’-DDT和p,p’-DDD可以很好的得到分离,但是针对17种或者22种组分,想要实现每种组分与其相邻峰基线分离,是比较困难的。
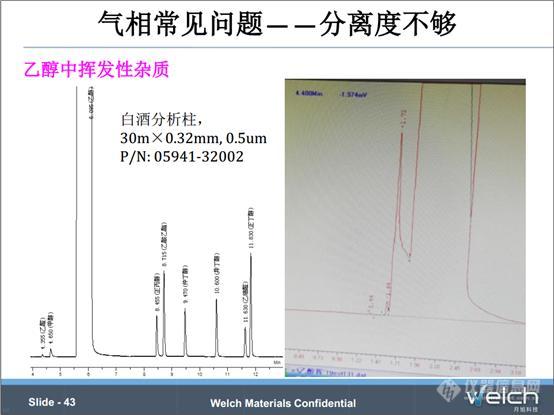
这个例子是2015版《中国药典》二部中乙醇项下挥发性杂质的检测,两张谱图用的是同一规格的色谱柱,即我们的白酒分析柱,30m×0.32mm×0.5μm,但是为什么会出现甲醇和乙醇的分离度差别如此之大的情况呢?
经具体了解,以前老师一直使用的是不锈钢填充柱,我们知道不锈钢填充柱的柱容量远远大于毛细管柱,所需要的载气流速也大于毛细管柱。所以换用毛细管柱的时候,需要减慢载气的流速,从而改善甲醇和乙醇的分离度。

下面就是我们看一下气相的两种通用型检测器,第一种就是氢火焰离子化检测器,也就是FID检测器。使用FID检测器,我们经常遇到的问题就是点不着火,针对这个问题,我们要分别从下边几点排除解决。
检查检测器的温度是否大于150℃。
观察点火线圈是否正常工作,点火时,如果点火线圈出现发红发亮的现象,就说明点火线圈没有损坏。
氢气和空气的实际比例是否达到1:10,可能有些老师会使用氢气发生器作为气源,氢气的纯度可能不够,也就是说氢气和空气的实际比例达不到1:10,此时需增加氢气流量。
氢气和空气走不同的路线到达喷嘴处,如果色谱柱在检测器的端口安装的位置太高,就阻碍了氢气的“道路”,导致氢气和空气不能正常相遇。所以色谱柱和检测器端口连接时,先将色谱柱插到底,然后往回抽1mm。
同样道路,喷嘴堵塞也导致氢气和空气不能相遇,此时需要清洗喷嘴。
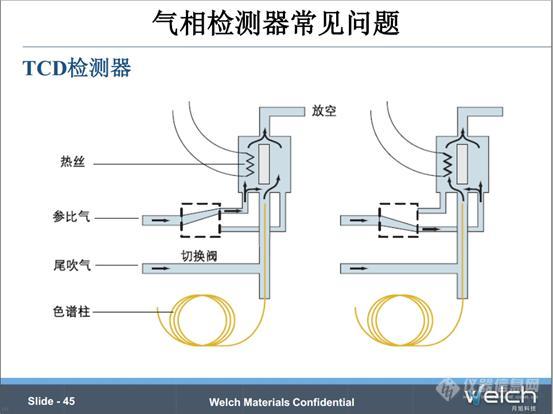
另一个应用比较广泛的通用型的检测器是TCD检测器。TCD检测器常见的一个问题是,没有通载气的情况下,先加了电流,导致热丝烧断,所以使用TCD检测器的时候一定要先通载气,然后再加电流。
气相柱老化时,不要接TCD检测器,以免色谱柱中的杂质污染TCD检测器。当然柱温不能超过色谱柱固定相的最高使用温度,避免固定液流失造成TCD检测器污染。


 题目里还是“气象”呢。
题目里还是“气象”呢。































