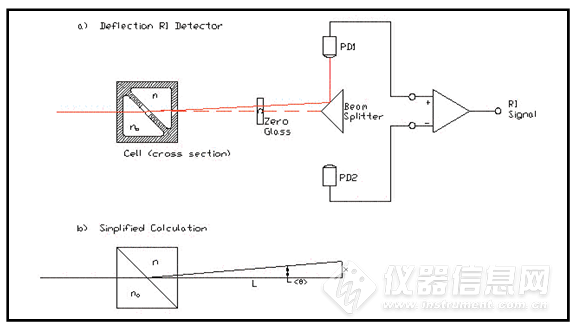
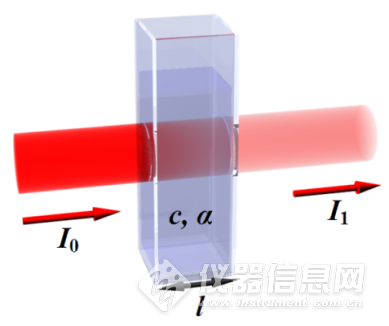
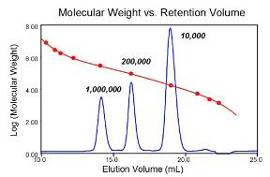
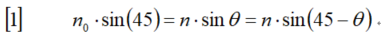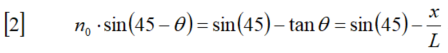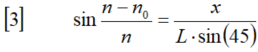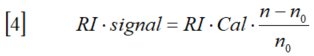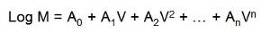新手上路
zyl3367898
第1楼2019/11/04
可以出系列丛书。
锁龙
第2楼2020/04/22
写的太好了,很有帮助。
附近的发货
第3楼2021/02/24
写的太有帮助了,感谢作者
品牌合作伙伴
执行举报