通标小菜鸟
第5楼2024/05/17
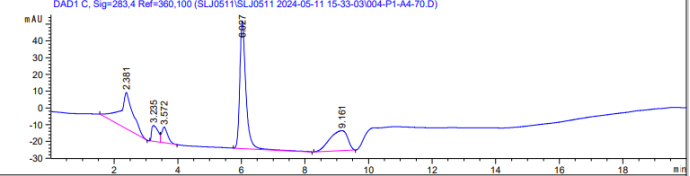
1、问题一:峰分叉的原因很有可能跟你进样浓度有关,你的谱图峰面积都3500以上了,那峰面积至少几万甚至几十万,这个浓度太高了,很容易出现分叉峰,建议控制浓度,或者减小进样量,改为2ul。另外你的色谱柱如果使用时间久了,柱效降低,也会造成分叉,但是我认为第一种可能性更大,你可以按照我说的方法试试,将进样量改为2ul,或者对样品进行稀释10倍。
2、问题二:其实你的出峰杂峰很少,不多,不像是基质带来的。而且你的纵坐标比例比较小,类似于方放大了,这种波动更像是流动相带来的,比如你设置了梯度洗脱,如果流动相组成比例发生改变,是会造成基线升高或者降低的,楼主可以尝试优化洗脱梯度,如果能分开,出峰好,改成等度也未尝不可。另外对于10min那没有出峰,那基本上就是没有,不过要看你样品是怎么前处理的,如果样品量小,也可能浓度太低测不到。

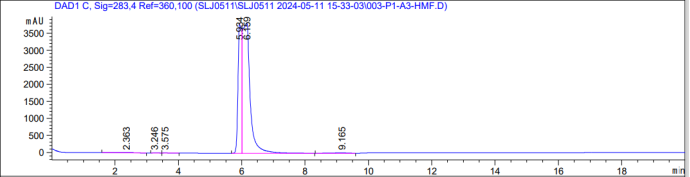 图一,羟甲基糠醛
图一,羟甲基糠醛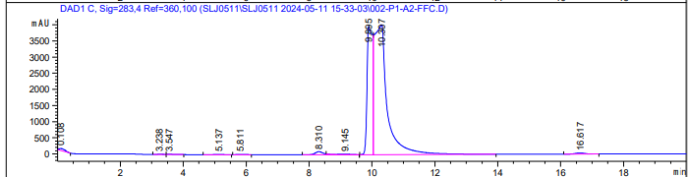 图二,糠
图二,糠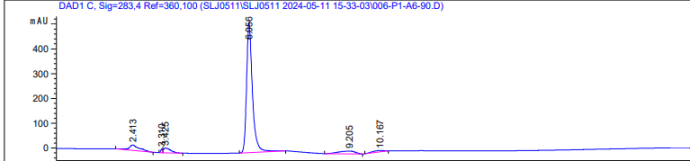 醛
醛