
Nature新成果|基于深度学习的糖肽碎片质谱预测
导读:方群教授团队开发了一种基于深度学习的模型用于预测完整糖肽的碎片谱图,并在多个数据集上进行了验证,该模型能够准确预测肽段和聚糖的碎片离子强度,并成功区分糖肽的聚糖异构体。
浙江大学求是特聘教授方群是化学系微分析系统研究所所长,国家杰出青年基金获得者。自1998年开始从事微流控芯片分析的研究工作。目前研究方向包括微流控液滴分析,微流控液相色谱、质谱和毛细管电泳分析,微型化分析系统研制,以及微流控系统在单细胞多组学分析、人工智能+化学、高通量筛选、微量生化分析、临床分析和现场分析中的应用。其团队在基于深度学习的糖肽碎片质谱预测方面取得重要成果,并发表在Nature Communications,方群教授为通讯作者。
液相色谱和串联质谱的联用(LC-MS/MS)是蛋白质组学和糖蛋白质组学研究中被广泛使用的方法,其核心是将碎片的谱图与候选肽段的理论或者实验谱图相匹配来鉴定肽段。目前所使用的大多数匹配方法是基于数据库搜索来实现的,其评分高低的依据是肽段或者糖肽的碎片离子的存在与否。但是这种搜索模式忽略了碎片离子的强度,作为一种补充的方法,基于谱图库的搜索会考虑碎片的存在和强度,从而产生更多的评分,并且可以应用于非数据依赖型的采集模式(DIA)。谱图库的数据来源除了实验,还可以通过预测的方式生成。目前,基于深度学习的方法已经在蛋白质组学当中得到了应用,可以预测蛋白酶酶切效率的可检测性,保留时间,离子淌度质谱中的碰撞截面积,MS/MS中的碎片离子强度,以及翻译后修饰的位点。预测谱库可以直接由蛋白质序列信息生成,并且基于特征评分模型已经可以区分真实信号和噪音。但是目前的方法仍旧无法预测完整糖肽的碎片谱图。
作者在传统的线性长短期记忆网络的基础上,引入了树结构的长短期记忆网络用于分析聚糖结构,并且利用带有注意机制的图形神经网络来分析糖肽的碎片化途径。具体地,糖肽在被输入模型之前被分为肽部分和聚糖部分,肽段部分分为序列和修饰,分别以独热编码和元素组成表示。聚糖部分则被表示为一颗由单糖为节点,糖苷键为边的树结构。然后分别采用两个线性的和两个树结构的长短期记忆网络来预测谱图,第一个线性的长短期记忆网络用于分析肽段的序列信息,第二个线性的长短期记忆网络用于分析肽段特征并预测b/y离子的强度。第一个树结构的长短期记忆网络则用于自下而上(非还原端到还原端)地分析聚糖结构,第二个树结构的长短期记忆网络则自上而下的分析聚糖特征。最后将肽部分和聚糖部分合并,得到糖肽的预测谱图。(如图1所示)
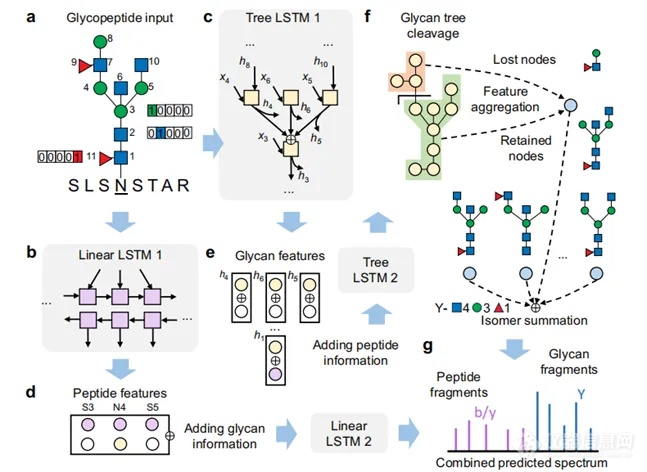
图1.糖肽碎片预测谱的模型 a输入的糖肽包括一个肽序列和一个聚糖树。b肽序列经线性长短期记忆(LSTM)网络处理。c通过树状LSTM网络遍历多糖树。d, e线性提取的肽特征与树状LSTM提取的聚糖特征相互融合。然后通过另一个线性LSTM网络对肽段特征进行处理,预测肽段b/y碎片的相对强度。糖的特征被另一个树状LSTM网络遍历,更新糖树中每个单糖节点的特征。f潜在裂解位点的特征由裂解后丢失或保留的单糖节点聚集而成。从相应的裂解中聚集结构特异性聚糖碎片的特征,以预测Y离子的相对强度,其中结构异构体碎片被组合。g肽和聚糖碎片离子最终合并形成输出的糖肽谱。
作者随后使用不同仪器设置的Orbitrap质谱获得的不同生物数据集对模型进行了训练和验证。在使用数据集进行训练之前,将其随机划分为三个子集,其中3/5用于拟合模型参数,1/5用于控制过拟合,剩下的1/5不参与训练(保留)用于性能估计。在Mouse 1和Human 1数据集上进行基准测试,用光谱角损失(SA)和点积(DP)作为判断的依据,碎片谱预测获得了非常高的相似性(图2a)。在训练集和保留集之间没有观察到实质性的指标差异,表明模型不是过拟合的。在此过程中发现,碰撞能量(CE)参数的设置相较于聚糖部分会对肽段部分产生更多的影响,因此在后续的过程中,还优化了CE参数的设置,并提高了预测的准确性(图2c,d)。用排除高甘露糖肽的数据集对带有分支的模型进行重新训练和验证,结果表明Y离子保持良好预测性能的情况下,B离子在不同生物和仪器设置下具有相当高的相似性,从而实现对整体的准确预测。
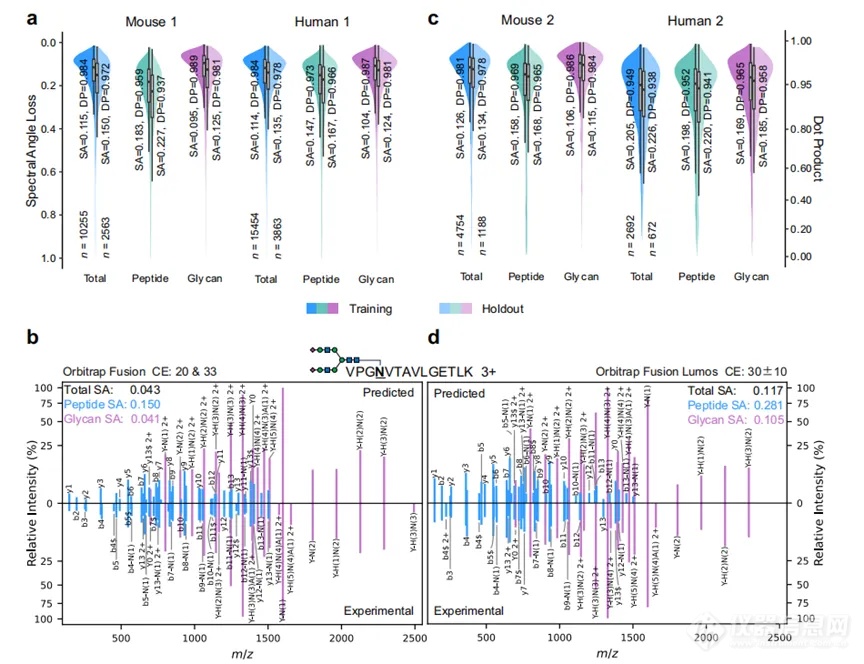
图2.预测性能评估 a Mouse1和Human1的训练集或保留集中所含糖肽的预测碎片离子强度与实验碎片离子强度之间的谱图相似性分布。b在Human1中,糖肽谱图匹配的镜像图比较了预测碎片强度和实验碎片强度。c模型微调后Mouse2和Human2的谱图相似度分布。d比较Human2中预测和实验碎片强度的镜像图。分别计算肽b/y离子和聚糖y离子的谱图相似性,以及肽和聚糖离子的总谱图相似性。
此外,作者探究了模型区分不同糖肽的聚糖异构体能力,从MS/MS数据集中选择非高甘露糖糖肽的谱图匹配作为查询谱图,进行谱库搜索。对于每个谱图匹配,候选糖肽是通过在预定义的聚糖空间中用其结构异构体替换原始聚糖来生成的。然后将查询谱与每个候选糖肽的预测谱进行比较,并计算它们之间的相似性度量(图3a)。图3b是从总共有超过1、2或3个候选谱图的情况中计算出正确鉴别被列为第一、第二或第三候选谱图的百分比。71%-80%的谱图匹配是正确的,并有92%-95%的概率能够在前三名的候选谱图中包含正确预测。此外还分别采用糖苷酶酶解和敲除的方式对末端的HexNAc和核心岩藻糖的鉴别进行了评估,结果如图3c,d所示。
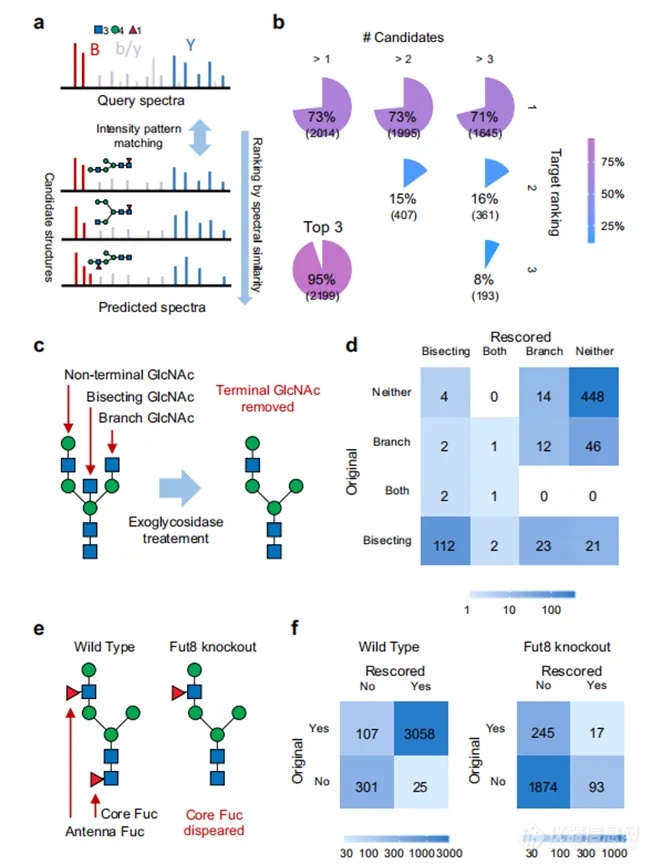
图3. 利用预测谱库区分结构异构体糖肽。a将查询谱与具有异构体聚糖结构的候选糖肽的预测谱进行比较,然后根据谱相似度评分对其进行排序。b标准糖肽数据集的候选排序结果。正确鉴别被列为第一、第二或第三候选的谱图的百分比由总共有超过1、2或3个候选的情况计算出来。前三名的图表显示了在有三个以上候选的案件中,正确鉴别在前三名候选中所占的百分比。c内糖苷酶处理末端HexNAc去除示意图。d利用预测谱库对内糖苷酶处理的小鼠脑数据集进行再分析,得到末端HexNAc识别的混淆矩阵。e在Fut8基因敲除小鼠中,核心岩藻糖消失,而在野生型小鼠中则保留。f通过重新分析Fut8基因敲除和野生型小鼠大脑数据集得出核心岩藻糖识别的混淆矩阵。
最后,作者将预测的谱图库与实验所得的DDA数据库分别用于DIA的数据检索,考察预测谱图是否能为DIA的数据检索带来更多的糖肽采集。在酵母的数据集分析中,与DDALib相比,预测谱库导致检测到的糖肽前体和位点特异性聚糖损失高达10%,但数据完整性略好。而在更加复杂的人类血清数据集中,预测的谱图库比DDA数据库所得的糖肽前体和位点特异性糖肽都更多(糖肽前体多7%,位点特异性糖肽多10%)。此外作者还生成了一个扩展的预测谱图库(PredExt),得到了更多的糖肽覆盖,在DIA的数据采集中也覆盖了更多的糖肽前体和位点特异性糖肽。并且对预测谱图库进行了精度的评估(图4d)。计算了不同混合比例样品间测得的糖肽丰度的倍数变化(图4e)。使用预测的谱库,与使用DDALib相比,人类糖肽丰度的倍数变化略高,而酵母糖肽的定量精度接近甚至有时优于DDALib。结果表明,预测谱库与实验谱库性能相当,适用于DIA数据分析。
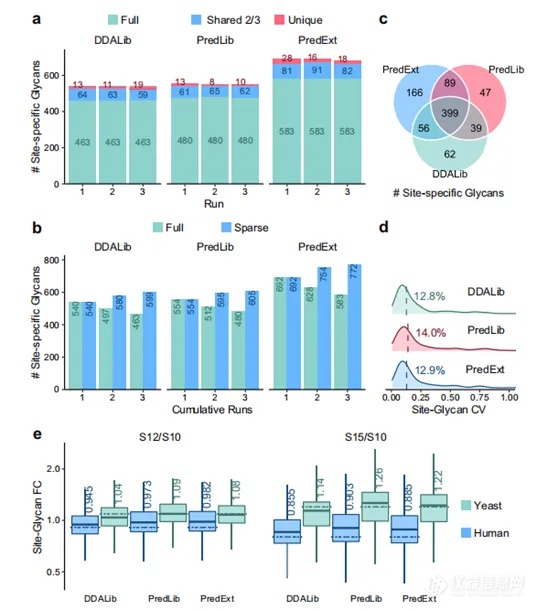
图4.DIA分析预测谱库的性能。a每次检测血清样本的鉴定数量。“full”表示在所有运行中观察到的识别;“shared2/3”表示在2次运行中观察到的识别;“unique”表示仅在一次运行中观察到的标识。b各批次血清样本的累积鉴定数。“full”表示在累积运行中共享的标识;“saprse”表示在累积运行中至少一次运行中观察到的标识。c使用不同文库shared2/3次血清样本的鉴定次数比较。d量化结果的变异系数(cv)。显示中位数。e混合生物样品定量结果倍数变化的箱形图可视化。根据每个样本三次重复的平均数量计算百分比变化。
来源于:仪器信息网


李惠琳课题组
总阅读量 0
相关阅读





1075万!西师范大学食品学院和西南科技大学分析测试中心仪器设备采购项目
dahua1981

仪器优选·液质联用仪
更多




相关会议
更多




热门评论
最新资讯
新闻专题
