推荐厂家
暂无
暂无
各位,现公司接到新的检测项目,要求检测纺织品中磷酸三(2,3-二溴丙基)酯 ,不知有没有哪位做, 用什么仪器方法进行检测?谢谢!
正在做萘的异丙基化反应,因为2,6-二异丙基萘和2,7-二异丙基萘标样太贵了,请教各位大牛,这两种物质在色谱中出峰顺序哪个在前?哪个在后? 非常感谢!!
我现在用液质做tepa三-(1-氮杂环丙基)氧化膦,可是买不到标准品,大家咋买到的啊,谁有用液质做的色谱图啊!http://images.basechem.org/struct/2011-03/ff30a0213c0d51d45505574710dd5862.gif





 留言咨询
留言咨询
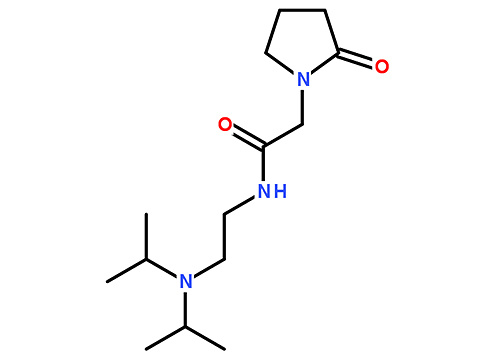 留言咨询
留言咨询
 400-860-5168转4940
留言咨询
400-860-5168转4940
留言咨询









 我要推广仪器
我要推广仪器
 下载APP
下载APP