推荐厂家
暂无
暂无

[align=center]缬沙坦清洁验证分析方法研究[/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]食品事业部:淮瑞娟[/align]试验方法:参照《中国药典》2010版(二部)中缬沙坦的分析方法。采用高效液相色谱法进行检测; 色谱柱为TC-C18(150×4.6mm,粒径:5um);流动相为乙腈-水-冰醋酸(500:500:1);检测波长为225nm。理论板数按缬沙坦峰计算不低于4000。缬沙坦峰与相邻杂质峰的分离度应大于1.5。试验结果:缬沙坦对照品溶液在0.1006μg/ml~5.03 μg/ml范围内呈良好的线性关系,r=1.0000,加标回收率可达到79.0%。结论:该方法操作简便、灵敏度高、专属性强,可用于缬沙坦清洁验证化学残留量的检测。[b]1、清洁验证方案提出的问题[/b]通过多种方法计算和考察,最终采用常规限度法作为缬沙坦设备清洁验证化学残留浓度的确定方法,通过比较另外两个原料药中的表面残留限度标准,最终确定27.0μg/dm2为目标残留物缬沙坦擦拭取样限度。中国药典2010版二部采用容量法测定缬沙坦的含量,而27.0μg/dm[sup]2[/sup]目标残留物限度容量法无法检测,因此考虑拟采用缬沙坦有关物质的方法——高效液相色谱法检测其化学残留物,但应进行分析方法学验证。同时考虑应做擦拭取样回收率考察擦拭取样方法的科学性和合理性2、系统适应性供试品溶液:取供试品50mg,精密称定,置100ml量瓶中,加流动相溶解并稀释至刻度,摇匀(0.5mg/ml)。对照溶液:精密量取1.0ml供试品溶液,置100ml量瓶中,加流动相稀释至刻度,摇匀;精密量取1.0ml置10ml量瓶中,加流动相稀释至刻度,摇匀(0.5ug/ml)。取供试品溶液和对照品溶液各10ul注入液相色谱仪,其中对照品溶液连续进样6次,记录色谱图。在供试品溶液获得的色谱图中,缬沙坦峰的理论踏板数应不小于4000。在对照溶液获得的色谱中,6次进样缬沙坦峰保留时间的RSD不大于1.0%,峰面积的RSD不大于5.0%。系统适用性检测结果[table][tr][td]测定次数[/td][td][align=center]1[/align][/td][td][align=center]2[/align][/td][td][align=center]3[/align][/td][td][align=center]4[/align][/td][td][align=center]5[/align][/td][td][align=center]6[/align][/td][/tr][tr][td]RT(min)[/td][td][align=center]6.288[/align][/td][td][align=center]6.288[/align][/td][td][align=center]6.288[/align][/td][td][align=center]6.288[/align][/td][td][align=center]6.288[/align][/td][td][align=center]6.288[/align][/td][/tr][tr][td]RSD%[/td][td=6,1][align=center]0%[/align][/td][/tr][tr][td]峰面积[/td][td][align=center]289385[/align][/td][td][align=center]289221[/align][/td][td][align=center]287769[/align][/td][td][align=center]286549[/align][/td][td][align=center]288336[/align][/td][td][align=center]287269[/align][/td][/tr][tr][td]RSD%[/td][td=6,1][align=center]0.39%[/align][/td][/tr][tr][td]理论塔板数[/td][td=6,1][align=center]10364[/align][/td][/tr][/table][align=center][img=,510,506]http://ng1.17img.cn/bbsfiles/images/2017/09/201709111135_01_2904018_3.png[/img][/align]对照品溶液进样图谱[b][b]3、线性关系[/b][/b]取缬沙坦对照品约25mg,精密称定,置50ml量瓶中,加流动相稀释至刻度,摇匀,精密量取1.0ml,置100ml量瓶中,加流动相稀释至刻度,摇匀得5ug/ml的缬沙坦对照品溶液。再精密量取本溶液,稀释得系列溶液(0.05~5ug/ml),分别取10ul进样测定,记录色谱峰及峰面积。以缬沙坦溶液浓度对峰面积进行线性回归计算,确认线性与范围。以标准溶液的浓度(ug/ml)为横坐标,其色谱峰面积为纵坐标绘制标准曲线。缬沙坦对照品溶液在0.1ug/ml至5ug/ml呈良好的线性关系,线性方程为:Y=536360.6440X﹣552.1797(r=1.0000)。结果见图线性关系图。[align=center][img=,554,378]http://ng1.17img.cn/bbsfiles/images/2017/09/201709111136_01_2904018_3.png[/img][img=,527,483]http://ng1.17img.cn/bbsfiles/images/2017/09/201709111136_02_2904018_3.png[/img][/align]对照品溶液浓度 0.1ug/ml进样图谱[b][b]4、定量限和检测限[/b][/b]4.1取线性范围测定项下线性最低浓度溶液(0.1ug/ml)10ul,注入色谱仪,记录色谱图,进样3次,信噪比S/N≧10即作为定量限浓度。对应检出限即为定量限。取定量限浓度溶液稀释三倍溶液10ul,注入色谱仪,记录色谱图,进样3次,信噪比S/N≧2即作为检测限浓度。对应检出限即为检测限。结果见表定量限和检测限结果,定量限为0.1006ug/ml,检测限为0.0335ug/ml。表 定量限和检测限结果[table][tr][td][align=center]峰名[/align][/td][td][align=center]峰面积[/align][/td][td][align=center]峰高[/align][/td][td][align=center]信燥比[/align][/td][td][align=center]信燥比平均值[/align][/td][td][align=center]定量限[/align][/td][/tr][tr][td=1,3][align=center]缬沙坦[/align][/td][td][align=center]56154[/align][/td][td][align=center]6144[/align][/td][td][align=center]11.6[/align][/td][td=1,3][align=center]11.9[/align][/td][td=1,3][align=center]0.1006μg/ml[/align][/td][/tr][tr][td][align=center]51299[/align][/td][td][align=center]6190[/align][/td][td][align=center]10.9[/align][/td][/tr][tr][td][align=center]55206[/align][/td][td][align=center]6343[/align][/td][td][align=center]13.2[/align][/td][/tr][tr][td=1,3][align=center]缬沙坦[/align][/td][td][align=center]19120[/align][/td][td][align=center]2383[/align][/td][td][align=center]2.9[/align][/td][td=1,3][align=center]5.2[/align][/td][td=1,3][align=center]0.0335μg/ml[/align][/td][/tr][tr][td][align=center]19988[/align][/td][td][align=center]2424[/align][/td][td][align=center]5.1[/align][/td][/tr][tr][td][align=center]20752[/align][/td][td][align=center]2509[/align][/td][td][align=center]7.6[/align][/td][/tr][/table][align=center][img=,554,502]http://ng1.17img.cn/bbsfiles/images/2017/09/201709111137_01_2904018_3.png[/img] [/align][align=center]定量限进样色谱图[/align][b][b]4.2精密度测定[/b][/b]取缬沙坦对照品约25mg,精密称定,置50mL量瓶中,加流动相稀释至刻度,摇匀,精密量取1.0ml,置100ml量瓶中,加流动相稀释至刻度,再精密量取1.0ml,置100ml量瓶中,加流动相稀释至刻度,摇匀得0.5μg/ml的缬沙坦对照品溶液。取对照溶液10μl注入液相色谱仪,重复进样6次,记录色谱图,并计算其相对标准偏差为0.9%,结果表明方法精密度良好精密度测定结果见表。表 精密度测定结果[table][tr][td]测定次数[/td][td]1[/td][td]2[/td][td]3[/td][td]4[/td][td]5[/td][td]6[/td][/tr][tr][td]峰面积[/td][td]299625[/td][td]309036[/td][td]305159[/td][td]303396[/td][td]298980[/td][td]302492[/td][/tr][tr][td]RSD%[/td][td=6,1][align=center]0.9%[/align][/td][/tr][/table][align=center][img=,554,534]http://ng1.17img.cn/bbsfiles/images/2017/09/201709111137_02_2904018_3.png[/img] [/align][align=center]精密度进样色谱图[/align][b][b]4.3中间精密度测定[/b][/b]另一分析员不同天按精密度测试方法连续制备6份对照溶液,并统计计算6+6次测定结果的RSD。取缬沙坦对照品约25mg,精密称定,置50mL量瓶中,加流动相稀释至刻度,摇匀,精密量取1.0ml,置100ml量瓶中,加流动相稀释至刻度,再精密量取1.0ml,置100ml量瓶中,加流动相稀释至刻度,摇匀得0.5μg/ml的缬沙坦对照品溶液。取对照溶液10μl注入另一台液相色谱仪,重复进样6次,记录色谱图,并计算其相对标准偏差为2.9%,结果表明方法中间精密度良好。表 中间精密度测定结果[table][tr][td]测定次数[/td][td]1[/td][td]2[/td][td]3[/td][td]4[/td][td]5[/td][td]6[/td][/tr][tr][td]峰面积[/td][td]299625[/td][td]309036[/td][td]305159[/td][td]303396[/td][td]298980[/td][td]302492[/td][/tr][tr][td]测定次数[/td][td][align=center]7[/align][/td][td][align=center]8[/align][/td][td][align=center]9[/align][/td][td][align=center]10[/align][/td][td][align=center]11[/align][/td][td][align=center]12[/align][/td][/tr][tr][td]峰面积[/td][td][align=center]305859[/align][/td][td][align=center]284112[/align][/td][td][align=center]289258[/align][/td][td][align=center]289604[/align][/td][td][align=center]284005[/align][/td][td][align=center]282539[/align][/td][/tr][tr][td]RSD%[/td][td=6,1][align=center]2.9%[/align][/td][/tr][/table][align=center][img=,577,453]http://ng1.17img.cn/bbsfiles/images/2017/09/201709111137_03_2904018_3.png[/img] [/align][align=center]中间精密度进样色谱图[/align][b][b]5.取样回收率[/b]5.1方法描述:一块1.5dm×1.5dm平整光洁的不锈钢板;用钢锥划出1dm×1dm的区域;配制5倍限度量的缬沙坦对照品溶液,精密量取1 ml溶液用注射器尽量均匀地涂布在1dm×1d mm的区域内,自然干燥或用电吹风温和地吹干不锈钢板;用无水乙醇润湿棉签,按以下取样方法擦拭不锈钢板,擦拭后棉签置于干燥洁净的具塞试管中,加塞,加入流动相适量,超声洗涤,使缬沙坦溶出;按高效液相检测法,计算回收率和RSD。[/b]5.2取样方法:将棉签头按在取样表面上,用力使其稍弯曲,平稳而缓慢地擦拭取样表面。在向前移动的同时将其从一边移到另一边。擦拭过程应覆盖整个表面。翻转棉签,让棉签的另一面也进行擦拭。但与前次擦拭移动方向垂直(如图)。擦拭完后,将棉签放入具塞试管,密封。另取一棉签重复操作一次。棉签擦拭取样示意图[align=center][img=,350,117]http://ng1.17img.cn/bbsfiles/images/2017/09/201709111138_01_2904018_3.png[/img][/align][align=center] 第一步 第二步[/align]影响因素:进行擦拭取样试验时应注意擦拭工具和不锈钢板的干扰。常用的擦拭工具为棉签,棉签易吸附残留物,其上面的隐形残留物也易被溶剂洗脱,但棉签易脱落纤维,故在使用前应用溶剂预先清洗,以免纤维遗留在取样表面;不锈钢板应平整光洁,清洗干净用电吹风吹干后再涂溶液,以免取样不均匀和引进杂质,而产生较大的RSD。还应注意取样方式,应尽可能采用固定的力度、擦拭速度和线路,以免产生较大的RSD。将不锈钢板清洗干净,同法重复三次。[b]5.3操作步骤[/b]准备一块1.5dm×1.5dm平整光洁的316L不锈钢板。分别在钢板上用钢锥划出1dm×1dm的区域。取缬沙坦对照品约27mg,精密称定,置200mL量瓶中,加流动相适量使溶解,并稀释至刻度,摇匀。精密量取1ml溶液(约5倍限度量),用注射器尽量均匀地涂布在1dm×1dm的区域内。自然干燥或用电吹风温和地吹干不锈钢板。取八支棉签,用无水乙醇浸泡棉签30分钟。取出棉签,挤压去除多余的乙醇,取其中的两支按取样方法擦拭不锈钢板。将擦拭后的两支棉签和两支空白棉签分别放入两支干燥洁净的具塞试管中,加塞,加入流动相约5ml,超声洗涤,重复洗涤三次。[b]5.4检验方法[/b]空白溶液制备:将三次空白棉签洗液合并转入50ml容量瓶中,加流动相稀释至刻度,用0.45μm的微孔滤膜过滤,即得。对照溶液制备:取缬沙坦对照品约25mg,精密称定,置50mL量瓶中,加流动相稀释至刻度,摇匀,精密量取1.0ml,置100ml量瓶中,加流动相稀释至刻度,摇匀,再精密量取1.0ml,置10ml量瓶中,加流动相稀释至刻度,摇匀,即得对照溶液。擦拭样品溶液制备:将三次擦拭洗液合并转入50ml容量瓶中,加流动相稀释至刻度,摇匀,即得擦拭样品溶液。测定法:分别精密量取空白溶液、对照溶液和供试品溶液10μl注入液相色谱仪,记录色谱图,空白溶液色谱图中杂质峰忽略不计,按外标法以峰面积计算。[b]5.5 结果计算测量值计算公式:测量值=50×200×CS×(Au/As)[/b] Cs-对照品溶液浓度,mg/ml As-对照品溶液峰面积 Au-擦拭样品溶液峰面积[align=center]按下式计算回收率:[img=,290,71]http://ng1.17img.cn/bbsfiles/images/2017/09/201709111139_01_2904018_3.png[/img][/align]结果见表,棉签擦拭取样方法检测结果。表明用棉签擦拭取样方法对清洁器具的取样回收率可达79.0%[align=center]表 棉签擦拭取样法检测结果[/align][table][tr][td][align=center]重复次数[/align][/td][td][align=center]试管编号[/align][/td][td][align=center]擦拭样品称样量(mg)[/align][/td][td][align=center]测量值(mg)[/align][/td][td][align=center]回收率(%)[/align][/td][/tr][tr][td][align=center]1[/align][/td][td][align=center]1[/align][/td][td][align=center]27.09[/align][/td][td][align=center]22.54[/align][/td][td][align=center]83.2[/align][/td][/tr][tr][td][align=center]2[/align][/td][td][align=center]2[/align][/td][td][align=center]27.09[/align][/td][td][align=center]21.40[/align][/td][td][align=center]79.0[/align][/td][/tr][tr][td][align=center]3[/align][/td][td][align=center]3[/align][/td][td][align=center]27.09[/align][/td][td][align=center]23.48[/align][/td][td][align=center]86.7[/align][/td][/tr][tr][td=2,1][align=center]回收率报告值[/align][/td][td=3,1][align=center]79.0%[/align][/td][/tr][tr][td=2,1][align=center]回收率RSD[/align][/td][td=3,1][align=center]4.6%[/align][/td][/tr][/table]
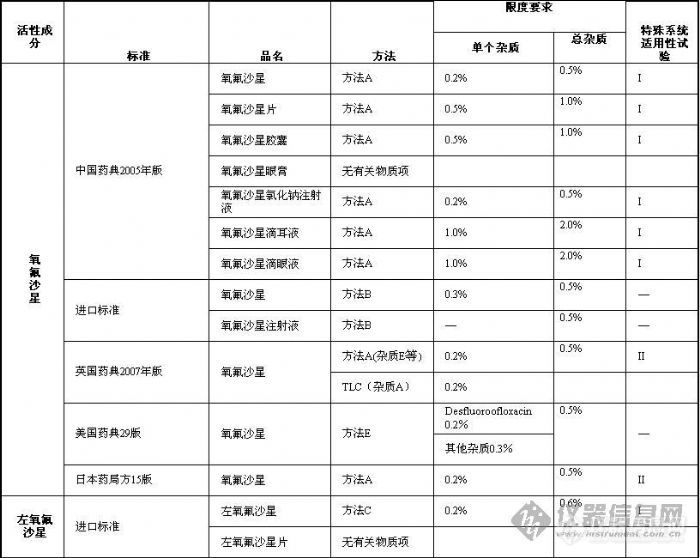
作者 张立雯 成海平 正文内容 【摘要】本文总结了国家标准中氧氟沙星、左氧氟沙星、盐酸左氧氟沙星、乳酸左氧氟沙星、甲磺酸左氧氟沙星系列药物的有关物质控制方法,分析了该类药物注册申报中有关物质控制存在的问题,希望能为研发者提供帮助。 【关键词】氧氟沙星、左氧氟沙星、有关物质一、概况 氧氟沙星(Ofloxacin)为合成的第三代广谱氟喹诺酮类抗菌药,对大多数革兰氏阳性菌和革兰氏阴性菌均有明显的抑制作用。临床上主要用于敏感菌所致的呼吸系统感染、泌尿生殖系统感染。氧氟沙星由日本第一制药株式会社研发,于1985年在日本、德国上市,制剂为口服片剂、注射剂等。目前国内已上市的氧氟沙星制剂有片剂、胶囊剂、颗粒剂、缓释制剂、小针、葡萄糖注射液和氯化钠注射液等。 左氧氟沙星(Levofloxacin)为氧氟沙星的左旋体,具有抗菌谱广、抗菌作用强的特点。日本第一制药株式会社于1993年在日本上市销售左氧氟沙星原料及片剂,并现已在英国、美国等多国上市。目前国内上市的左氧氟沙星制剂主要有片剂、小针、葡萄糖注射液和滴眼剂等。另外,国内已批准上市的左氧氟沙星还有其盐酸盐、乳酸盐和甲磺酸盐,三种加酸根的左氧氟沙星均有片剂、胶囊剂、注射制剂等多种剂型上市。二、国家标准中有关物质控制方法比较 氧氟沙星系列药物的有关物质测定国家标准大多采用HPLC法,列表比较见表1。 表1 氧氟沙星系列药物的有关物质测定方法与限度的比较 这些方法有很多相似的地方,如均采用ODS柱,色谱条件与含量测定色谱条件相同,按照主成分自身稀释对照法定量等。但也有一些不同的地方值得关注,作者从以下三个方面来对这些国家标准方法的不同之处进行比较。1、流动相 按照流动相的不同,作者将有关物质测定方法分为六种,具体如下: 方法A:醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH至 2.2)-乙腈(85∶15)为流动相,在294nm下检测; 方法B:略; 方法C:略; 方法D:己烷磺酸钠[取己烷磺酸钠0.98g,加磷酸盐缓冲溶液(取磷酸二氢钾6.8g,加水溶解并稀释至 1000ml,加0.05mol/L磷酸约500ml,使pH为2.4)]-甲醇(3∶1)为流动相,在293nm下检测; 方法E:磷酸缓冲溶液(溶解27.2磷酸二氢钾在1000ml水中,用磷酸调节pH至2.4)-乙腈(90∶10)为流动相,在294nm下检测; 方法F:己烷磺酸钠[取己烷磺酸钠0.98g,加磷酸盐缓冲溶液(取磷酸二氢钾6.8g,加水溶解并稀释至 1000ml,加0.05mol/L磷酸约500ml,使pH为2.4)]-甲醇(65∶35)为流动相,在230nm下检测。 六种流动相的共同特点是:组成均是酸性缓冲溶液加有机溶剂(甲醇或乙腈)。方法A、B、E未加表面活性剂,方法C、D加有表面活性剂己烷磺酸钠。除方法F在230nm下检测外,其他方法均在294或293nm下检测。2、主要杂质 英国药典收载了氧氟沙星杂质A、B、C、D、E、F共6个已知杂质,依次分别为去哌嗪环、去羧基、去氟、氟取代位置不同、去甲基以及氮氧化的化合物物。英国药典氧氟沙星原料药采用TLC法控制杂质A,采用HPLC法控制其他已知和未知杂质。美国药典重点关注了杂质desfluoroofloxacin,日本药局方重点关注了ofloxacin demethyl substance,均与英国药典的杂质E相同,是氧氟沙星的去甲基化合物,该化合物为氧氟沙星的主要降解产物,光照下极易产生。美国药典给出了desfluoroofloxacin的相应因子为1.13。中国药典没有明确已知杂质,但有关物质检查时采用以下光照降解法进行特殊系统适用性试验,光照试验中产生的杂质即为氧氟沙星、左氧氟沙星的主要杂质。 美国药典中desfluoroofloxacin按加校正因子的主成分自身稀释对照法定量,美国药典氧氟沙星其他杂质和其他国家标准中氧氟沙星或左氧氟沙星所有杂质按不加校正因子的主成分自身稀释对照法定量。有关物质限度的要求详见表1。3、特殊系统适用性试验 氧氟沙星、左氧氟沙星及其盐的含量测定和有关物质检查方法的系统适用性试验除通常的进样精密度、记录时间、理论塔板数等的要求外尚有一项较为特殊的系统适用性试验,其他标准方法采用I法,日本药局方采用II法,详述如下: 特殊系统适用性试验I(光照降解法):取供试品溶液于无色试管中,用日光灯(2500lux或3500lux)或紫外灯(254nm)照射1小时或3小时或4小时,取此液注入液相色谱仪,记录色谱图,相对保留时间约为主峰1.2处应能检测出色谱峰。 特殊系统适用性试验II(杂质对照品法):氧氟沙星和杂质E(ofloxacin impurity E CRS,英国药典)或氧氟沙星的去甲基物(ofloxacin demethyl substance)分离度不得低于2.0或2.5。 由于缺少杂质对照品,国内氧氟沙星系列药物的有关物质测定系统试验常常是采用光照降解法。也正是因为缺少杂质对照品,系统适用性试验才显得尤为重要,是考察系统分离能力的重要指标。 另外,左氧氟沙星及其盐的原料和制剂均需检查右旋异构体,方法基本相同,均是采用硫酸铜-L异亮氨酸溶液-甲醇或硫酸铜-D苯丙氨酸溶液-甲醇为手性流动相检测,限度要求不得过0.8%或1.0%。在此就不详加讨论。三、注册申报中存在问题与探讨1、不重视系统适用性试验 氧氟沙星系列药物的特殊系统适用性试验常常被忽视,其实却非常重要。若不做该项试验,就不能保证所采用的系统能将最难分离的相对保留时间1.2倍的色谱峰分离出来,就有可能得到错误的结果。 审评中曾发现申报盐酸左氧氟沙星注射液的某厂家自测盐酸左氧氟沙星含量较药检所检验结果高约5%(含量测定色谱条件与有关物质检查一致)。仔细审查其图谱,发现未按盐酸左氧氟沙星注射液的已有国家标准用光照降解法进行系统适用性试验,且色谱峰明显拖尾。其测定结果偏高很可能是紧随主峰之后的杂质峰包裹进了主峰。 申请人往往会留意进样精密度、理论塔板数这样的常规系统适用性试验,却常常忽略了光照降解系统适用性试验,此种现象在申报资料中占很大比例。究其原因,是试验人员没有理解到此项系统适用性试验的目的和重要性,希望提醒申请人提高对系统适用性试验的重视程度。2、没有杂质个数与含量的详细对比 申报资料中杂质对比研究通常的做法就是按照国家标准方法检验一下自制品和已上市对照药品,若都在标准规定范围内,就认为自制品与已上市药品质量相当。其实这样的做法是对杂质对比的目的和比什么不甚明了的表现。杂质对比一方面要了解自己的产品与已上市品杂质有哪些不同,另一方面要了解制剂过程中有没有新产生的杂质,若有新产生的杂质,应加以控制。所以比较就要落到列表对比杂质的个数与含量上,泛泛地比较杂质总量是不足以说明问题的。 例如,申报盐酸左氧氟沙星、乳酸左氧氟沙星的注射剂采用方法D测定的较为多见。在该色谱条件下,相对保留时间为0.23、0.43、1.2左右的杂质峰较常见,其中相对保留时间为1.2的色谱峰是稳定试验中含量有所增加的主要杂质。 另外,统计杂质的个数时要注意“忽略限度”。英国药典氧氟沙星有关物质项下明确规定:Disregard any peak with an area less than 0.1 times the area of the principal peak in the chromatogram obtained with reference solution (a),即忽略面积小于对照溶液主峰面积0.1倍的色谱峰(0.02%)。中国药典没有这么详细的规定,但从实际操作来看,为增强方法的严谨性和数据的可比性,建议申请人在统计杂质个数时应明确忽略限度。3、强力破坏试验降解程度不合适 有关物质检查方法学验证的重要项目就是通过强力破坏试验考察方法的专属性,但强力破坏试验破坏程度的掌握不尽合理。较容易出现的情况是破坏太轻微,酸、碱、氧化、光照破坏均几乎未产生可检测的杂质,这样就无法判断所采用的色谱条件分离能力是否符合要求,破坏试验失去意义。另一种极端是破坏过度,主峰降解了大半,产生大量重叠的杂质色谱峰,这样很难找到合适的色谱条件将所有的降解产物分离。个人认为,适度的破坏应是采用比贮藏中可能遇到的最强条件稍强烈的条件降解,产生比贮藏中可能产生的稍多杂质,若所选色谱条件能将这些杂质都分离,就是专属性符合要求的。 以上问题是氧氟沙星系列药物审评中经常遇到的问题,希望能为研发者提供帮助,共同努力提高我国仿制药的研发水平。 [img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907202227_160694_1612824_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/07/200907202231_160696_1612824_3.jpg[/img]
做缬沙坦很久了,一直用柏金柱,分离度和峰形都很好,但是最近在溶剂峰后面1min出了两个杂质,柏金柱分不开。1. 我看到思博尔色谱柱的介绍说能分离结构相似化合物,我猜测这两个杂质应该是结构相似的,不知道思博尔能不能分开?2. 不知道缬沙坦会不会在思博尔上拖尾?3. 思博尔有两个型号:C18和C18-EP,不知道哪个更适合我这种情况?求版内高手相助,谢谢!!







 400-805-8969
400-805-8969
 留言咨询
留言咨询
 留言咨询
留言咨询
 400-860-5168转4406
留言咨询
400-860-5168转4406
留言咨询









 我要推广仪器
我要推广仪器
 下载APP
下载APP


