推荐厂家
暂无
暂无
在进行苦参鉴别(3)(4)时,用的是混合对照品,对照品用的是乙醇配制得,而供试品用的则是三氯甲烷,点板之后对照品没有出现斑点。在给对照品用三氯甲烷重新配置后,出现斑点,与样品可以出现相同的斑点。请问是不是药典错了啊
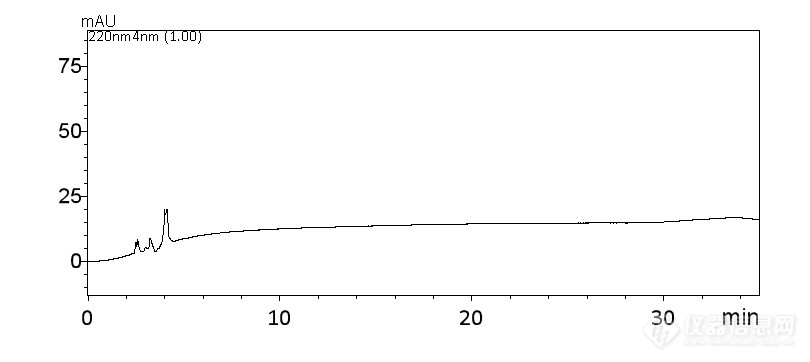
[align=center][b]流动相混合方式对苦参碱及氧化苦参碱出峰影响[/b][/align][b]1. 色谱条件与系统适用性试验[/b] 色谱柱Agilent ZORBAX NH[sub]2[/sub](Agilent ZORBAX SB C18);以乙腈-异丙醇- 3 % 磷酸溶液(80 :5 : 15)为流动相 ;检测波长为210nm。理论板数按氧化苦参碱峰计算应不低于4000。[b]2. 对照品溶液的制备[/b] 取苦参碱对照品、氧化苦参碱对照品 适量,精密称定,加流动相分别制 成苦参碱94.74 μg/ml, 氧化苦参碱156.65μg/ml的混合溶液,即得。[b]3. 供试品溶液制备[/b] 取本品粉末(过三号筛)约0. 3g,精密称定,置具塞锥形瓶中,加浓氨试液0. 5ml,精密加入三氯甲 烷20ml,密塞,称定重量,超声处理 (功率250W,频率33k Hz)30分钟,放冷,再称定重量,用三氯甲烷补足减失的重量,摇匀,滤过,精密量取续滤液5ml,加在中性氧化铝柱 (100〜 200目,5g,内径 1 cm )上 ,依次以三氯甲烷、三氯甲烷 -甲醇(7 : 3)混合溶液各20ml洗脱,合并收集洗脱液,回收溶剂至干,残渣加无水乙醇适量使溶解,转移至10ml量 瓶中,加无水乙醇至刻度,摇匀,即得。[b]4. 样品分析[/b] 依次将混合对照品溶液和供试品溶液进样10 μL,记录苦参碱、氧化苦参碱保留时间及峰面积。[b]5. 结果[/b]5.1 以乙腈(A)-异丙醇(B)- 3 % 磷酸溶液(C)三相流动相泵前混合 混合对照品溶液及供试品溶液色谱图分别见图1、图2.[img=,690,308]https://ng1.17img.cn/bbsfiles/images/2019/07/201907041713523978_9340_2613952_3.jpg!w690x308.jpg[/img] 图1 对照品溶液色谱图[img=,690,308]https://ng1.17img.cn/bbsfiles/images/2019/07/201907041714189908_9161_2613952_3.jpg!w690x308.jpg[/img] 图2 供试品溶液色谱图5.2 以乙腈-异丙醇-3%磷酸溶液(80:5:15)混合溶液为流动相 分别将乙腈、异丙醇、3%磷酸溶液过膜,之后将乙腈-异丙醇-3%磷酸溶液按照药典比例混合均匀于同一流动相瓶中,超声脱气,在线脱气。混合对照品溶液及供试品溶液色谱图见图3、图4。[align=left][img=,690,308]https://ng1.17img.cn/bbsfiles/images/2019/07/201907041714538638_4360_2613952_3.jpg!w690x308.jpg[/img] [/align] 图3对照品溶液色谱图[img=,690,308]https://ng1.17img.cn/bbsfiles/images/2019/07/201907041715257979_1578_2613952_3.jpg!w690x308.jpg[/img] 图4 供试品溶液色谱图[b]6 讨论[/b] 以乙腈(A)-异丙醇(B)- 3 %磷酸溶液(C)三相流动相在四元泵上泵前混合,或者以乙腈为流动相A、异丙醇-3%磷酸溶液(5:15)为流动相B二元高压泵上泵后混合,对照品溶液和供试品溶液苦参碱、氧化苦参碱出峰异常,见图1、图2。只有预先将乙腈-异丙醇-3%磷酸溶液按照药典(80:5:15)比例预先混合均匀,脱气,对供试品溶液及对照品溶液梯度洗脱,苦参碱和氧化苦参碱出峰正常,见图3、图4。
黄连味苦性寒,具有清热燥湿、泻火解毒的功效。《中国药典》2020年版规定黄连为毛茛科黄连属植物黄连Coptis chinensis Franch.、三角叶黄连C. deltoidea C. Y. Cheng et Hsiao或云连C. teeta Wall.的干燥根茎。以上3种分别习称“味连”“雅连”“云连”,经课题组前期调研以味连产量最多,主产于我国重庆、湖北、四川等地[1]。现代研究表明,黄连含有多种活性成分,可发挥多种药理作用[2],包括抗炎、抗病毒、抗菌、抗癌、镇痛、抗抑郁、降血糖等作用,临床应用极广[3]。苦参性寒、味苦,为豆科苦参属植物苦参Sophora flavescens Ait.的干燥根,主产于我国内蒙古、河南、山东、安徽等地[1],具有抗菌、抗肿瘤、镇痛、抗炎、防治心力衰竭、心律失常及心肌缺血等多种功效[4-5]。 现代研究表明,生物碱类化合物是黄连及苦参的主要活性成分。苦参碱、氧化苦参碱可发挥抗炎、镇痛效果[6-7],其机制可能与降低促炎因子,升高抗炎因子有关;氧化苦参碱、苦参碱也可发挥抗肿瘤作用,其机制可能与抑制癌症基因表达,促进肿瘤细胞凋亡,抑制肿瘤细胞生长有关[8];而苦参碱、氧化苦参碱、槐定碱也可对多种菌株具有一定的抑菌作用[9]。木兰花碱可通过活性氧(reactive oxygen species,ROS)/鼠类肉瘤病毒癌基因(Kirsten rat sarcoma viral oncogene,KRAS)/单磷酸腺苷活化蛋白激酶(adenosine monophosphate activated protein kinase,AMPK)通路抑制结直肠癌SW480细胞的增殖和有氧糖酵解,从而发挥对结直肠癌的治疗效果[10];药根碱、巴马汀、表小檗碱、黄连碱、小檗碱可联合发挥降糖作用[11],其效果可能与调控丝氨酸-苏氨酸激酶1(serine/threonine kinase 1,LKB1)/ AMPK/CREB分子调节转录共激活剂2(CREB-regulated transcription coactivator 2,TORC2)信号通路抑制肝脏糖异生等有关[12];小檗碱具有抗炎作用,可保护螺旋神经节细胞免受巨细胞病毒诱导的凋亡作用,其机制与通过途径抑制线粒体活性氧的产生有关[13]。 除此之外,小檗碱、表小檗碱、巴马汀等生物碱类成分也可联合发挥抗心律失常作用[14]。基于此,选择苦参中苦参碱、槐定碱、氧化苦参碱及黄连中木兰花碱、非洲防己碱、药根碱、表小檗碱、黄连碱、巴马汀、小檗碱来作为黄连-苦参药对的代表性药效成分,用于研究该类成分溶出量与药对配比的关系。药对作为中药配伍的最小单元,是复方研究的重要组成部分之一[15]。用于不同疾病的治疗时,不同量的配比会有不同效果的相关呈现,因此,首先需要对黄连-苦参药对配比的不同物质基础,即量-质[16]相关性进行剖析比较,为进行量-效[17]相关性提供依据,为临床合理配比提供参考[18]。 1 仪器与试药 1.1 主要仪器 Waters e2695型高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]系统,Waters 2998型二极管阵列检测器(PDA),美国Waters公司;BBA224S-CW型电子天平,赛多利斯科学仪器(北京)有限公司;TGL-16C型离心机,上海安亭科学仪器厂;EPED-E2-20TS型超纯水一体机系统,南京易普易达科技发展有限公司;GM-0.5B型真空泵,天津市津腾实验设备有限公司;KH-500V型超声器,昆山禾创超声仪器有限公司。 1.2 药品及试剂 1.2.1 药材与饮片 本研究所选择黄连(产地重庆石柱黄水,批号20230411)及苦参(产地内蒙古赤峰市,批号2020121604)药材,均经南京中医药大学药学院刘圣金教授鉴定,分别为毛茛科黄连属植物黄连C. chinensis Franch.的干燥根茎和豆科苦参属植物苦参S. flavescens Ait.的干燥根。 1.2.2 对照品 表小檗碱(批号J24HB186173)、盐酸小檗碱(批号S01A10K94340)、盐酸黄连碱(批号T21S11C125202)、药根碱(批号D18GB171805)、盐酸巴马汀(批号Z16J10X79792)、非洲防己碱(批号W14J8Z37548)、木兰花碱(批号R21M9F61834)、苦参碱(批号M14GB141405)、氧化苦参碱(批号G14N11KL130769)、槐定碱(批号F18F7S9784),HPLC质量分数均≥98%,均购自上海源叶生物科技有限公司。 1.2.3 试剂 乙腈、甲醇,色谱纯,安徽天地高纯溶剂有限公司;磷酸、盐酸、无水乙醇,分析纯,国药集团化学试剂有限公司;纯净水,屈臣氏集团(香港)有限公司;磷酸二氢钾,分析纯,南京化学试剂股份有限公司。 2 方法与结果 2.1 不同配比黄连-苦参药对指纹图谱的建立 2.1.1 色谱条件 色谱柱为Venusil XBP C18(2)(250 mm×4.6 mm,5 μm);柱温30 ℃;体积流量0.8 mL/min;流动相为乙腈-3 g/L磷酸二氢钾溶液(加入200 μL磷酸调节pH值),梯度洗脱:0~10 min,10%乙腈;10~25 min,10%~24%乙腈;25~35 min,24%乙腈;35~60 min,24%~35%乙腈;60~62 min,35%~60%乙腈;62~65 min,60%~10%乙腈;65~70 min,10%乙腈;分析时间70 min,进样量10 μL;检测波长220 nm。 2.1.2 混合对照品溶液的制备 取非洲防己碱、药根碱、表小檗碱、盐酸小檗碱、盐酸巴马汀、盐酸黄连碱、木兰花碱、苦参碱、氧化苦参碱、槐定碱对照品各适量,分别置于10 mL量瓶中,加甲醇溶解并定容,即得各对照品储备液。分别取适量上述11种对照品储备液,置于同一10 mL量瓶中,加甲醇稀释并定容,制得上述成分质量浓度分别为0.20、0.16、0.24、0.25、0.25、0.44、0.26、0.21、0.80、0.36 mg/mL的混合对照品溶液。 2.1.3 供试品溶液的制备 制备黄连药材粉末(过二号筛)及苦参药材粉末(过三号筛),将上述黄连及苦参依照5∶1、4∶1、3∶1、2∶1、1∶1、1∶2、1∶3、1∶4、1∶5共9个质量比例,进行称取后分别充分混合,并称取单一黄连药材粉末及单一苦参药材粉末作为对照药材,各比例药对总质量及单一药材质量均为12 g。每个比例平行称取各药对3份,将药对以10倍量水浸泡0.5 h后,煎煮1.5 h,取1次滤液;将滤渣加入8倍量水煎煮1.5 h,取2次滤液。将2次滤液混合后抽滤,12 000 r/min离心(离心半径10.4 cm)10 min,取上清液,取1 mL上清液加入4 mL甲醇,以0.45 μm微孔滤膜滤过,即得供试品溶液。 2.1.4 精密度试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样测定6次,考察特征峰的保留时间和峰面积一致性。以盐酸小檗碱的保留时间和峰面积为参照分别计算相对保留时间及相对峰面积。计算得各共有峰相对保留时间的RSD<0.20%,相对峰面积的RSD<2.13%,结果表明仪器精密度良好。 2.1.5 稳定性试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件每隔4 h进样1次,共测定24 h,考察特征峰保留时间和峰面积的一致性。以盐酸小檗碱的保留时间和峰面积为参照分别计算相对保留时间及相对峰面积。计算得各共有峰相对保留时间的RSD<0.21%、相对峰面积的RSD<2.46%,结果表明该供试品溶液在室温放置24 h内稳定性良好。 2.1.6 重复性试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,平行制6份,按照“2.1.3”项下方法制备供试品溶液,分别按“2.1.1”项下色谱条件进样分析,考察特征峰保留时间和峰面积的一致性。以盐酸小檗碱的保留时间和峰面积为参照分别计算相对保留时间及相对峰面积。计算得各共有峰相对保留时间的RSD<0.18%、相对峰面积的RSD<1.57%,表明该方法重复性较好。 2.1.7 黄连-苦参药对指纹图谱的建立及相似度评价分析 将黄连及苦参药材依照“2.1.3”项下方法制备成供试品溶液(S1~S9依次为黄连-苦参比例为5∶1、4∶1、3∶1、2∶1、1∶1、1∶2、1∶3、1∶4、1∶5),再按“2.1.1”项下色谱条件进样分析,记录色谱图。将图谱输入《中药色谱指纹图谱相似度评价系统(2012版)》,设置编号S7的样品(黄连-苦参为1∶3)图谱为参照,采取中位数法[19],将时间窗宽度设置为0.1 s,进行多点校正,建立黄连-苦参药对的HPLC指纹图谱和对照指纹图谱(R,图1),指认9批黄连-苦参药对的16个共有峰。采用《中药色谱指纹图谱相似度评价系统(2012版)》对9批黄连-苦参药对进行相似度评价[20]。结果显示,9批黄连-苦参药对和R之间的相似度均大于0.95,这表明各批次黄连-苦参药对的相似性较好,整体质量稳定,可以用于考察黄连-苦参药对水煎液。以分离度较好、峰面积较大的小檗碱(峰16)为参照峰(S),得到9批黄连-苦参药对16个共有峰相对保留时间的RSD为0.175%~0.894%,提示各批次黄连-苦参药对共有峰的保留时间稳定 2.1.8 黄连-苦参药对指纹图谱色谱峰归属认定 通过比对单味药的色谱峰[21],不同比例配伍黄连-苦参药对HPLC指纹图谱16个共有峰中峰2~6号共5个峰均来源于单味药苦参,峰1、7~16号共11个峰来源于单味药黄连(图1)。通过对比混合对照品溶液色谱图(图2)及黄连、苦参及样品HPLC叠加图(图2)对各样品指纹图谱的各峰进行定性认证[22],得到2、3、6号峰分别为苦参碱、槐定碱、氧化苦参碱,属于单味药苦参;8、11~16号峰分别为木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱,属于单味药黄连。 2.1.9 黄连-苦参药对各共有峰相对峰面积差异分析 将各比例药对中黄连-苦参药对生药量以黄连、苦参单煎样品的生药量为标准,换算成一致的量,并以黄连及苦参单煎样品峰面积作为参比,比较不同配比黄连-苦参药对的共有峰相对峰面积,结果见表2。可知在不同程度配比下,各共有峰相对峰面积均有不同程度的变化,绝大部分表现出显著性差异。除属黄连药材的10、13号峰各相对峰面积相比药材单提均有所下降外,其余峰均表现为升高,表明配比后成分的溶出对苦参总体表现为促进作用,而对黄连的不同成分表现为促进和抑制的不同作用。1、5、7号峰在黄连-苦参为2∶1时相对峰面积最大;2~4、8、10号峰在黄连-苦参为5∶1时相对峰面积最大;11~16号峰在黄连-苦参为4∶1时相对峰面积最大;6号峰在黄连-苦参为1∶3时相对峰面积最大;9号峰在黄连-苦参为3∶1时相对峰面积最大,提示在方剂中使用不同配比黄连-苦参药对治疗疾病,可能与不同配比下药对中成分的溶出变化有关[23]。 2.2 不同配比黄连-苦参药对中差异性成分含量测定 2.2.1 色谱条件 按照“2.1.1”项下色谱条件进行测定。设定在波长为205 nm时,对苦参碱、槐定碱、氧化苦参碱进行测定;在波长为220 nm时,对木兰花碱进行测定;345 nm时,对非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱进行测定。此时各指标性成分均为最大吸收波长。 2.2.2 混合对照品溶液的制备 依照“2.1.2”项下方法制备混合对照品溶液。 2.2.3 供试品溶液的制备 依照“2.1.3”项下方法制备9个比例的黄连-苦参药对供试品溶液,每个比例制备3个供试品溶液作为平行对照。 2.2.4 线性关系考察及检测限、定量限 对照品母液的配制:取苦参碱、槐定碱、氧化槐果碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱对照品各适量,分别置于10 mL量瓶中,加甲醇溶解并定容,制得上述成分质量浓度分别为0.98、0.40、0.85、0.35、0.31、0.35、0.36、0.36、0.35、0.81 mg/mL的对照品溶液。 取各对照品母液,逐级稀释0、2、4、8、16、32、64倍,按照“2.1.1”项下色谱条件进行测定。以各差异性成分的质量浓度为横坐标(X)、峰面积为纵坐标(Y)绘制标准曲线,进行线性回归,得回归方程,结果见表3,表明各成分线性关系良好。 依照信噪比,即S/N为3∶1及S/N为10∶1对各成分的检测限及定量限进行检测,结果见表3。 2.2.5 精密度试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法制备供试品溶液,按“2.1.1”项下色谱条件连续进样6次,记录各差异性成分的峰面积。结果显示,苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱峰面积的RSD分别为1.40%、2.13%、1.37%、2.11%、0.91%、0.69%、1.25%、1.19%、0.17%、0.14%,结果表明仪器精密度良好。 2.2.6 稳定性试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法制备供试品溶液,于室温放置0、4、8、12、16、20、24 h,按“2.1.1”项下色谱条件进样分析,记录各差异性成分的峰面积。结果显示,苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱峰面积的RSD分别为1.57%、2.24%、2.22%、2.46%、0.22%、0.16%、0.65%、0.05%、0.14%、0.20%,表明各差异性成分在室温放置24 h内稳定性较好。 2.2.7 重复性试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,按照“2.1.3”项下方法平行制备供试品溶液6份,再按“2.1.1”项下色谱条件进样分析,记录各差异性成分的峰面积,并根据标准曲线计算含量。结果显示,苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱质量分数的RSD分别为1.16%、1.24%、1.33%、1.57%、1.05%、1.19%、1.42%、1.30%、1.21%、1.22%,表明该方法重复性良好。 2.2.8 加样回收率试验 依照黄连与苦参比例1∶3,精密称取黄连药材粉末3 g及苦参药材粉末9 g,平行称取6份,分别加入含有苦参碱0.31 mg、槐定碱0.20 mg、氧化苦参碱1.52 mg、木兰花碱0.07 mg、非洲防己碱0.08 mg、表小檗碱0.27 mg、药根碱0.06 mg、黄连碱0.22 mg、巴马汀0.21 mg、小檗碱0.79 mg的对照品溶液5 mL,按照“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样分析,记录各标志性成分的峰面积,并计算平均加样回收率。结果显示,苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱的平均加样回收率分别为100.2%、100.1%、100.3%、100.2%、101.2%、100.7%、99.8%、101.1%、100.60%、101.0%,RSD分别为0.67%、0.97%、0.89%、0.97%、0.56%、0.70%、0.57%、0.71%、0.99%、0.85%,表明该方法准确度良好。 2.2.9 不同配比黄连-苦参药对水煎液成分含量测定及比较 取9个不同比例的黄连-苦参药对药材粉末,精密称定,按照“2.1.3”项下方法制备供试品溶液,再按“2.1.1”项下色谱条件进样分析,记录各差异性成分的峰面积,并根据标准曲线计算苦参碱、槐定碱、氧化苦参碱、木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱的含量。将各比例药对中黄连-苦参药对生药量以黄连、苦参单煎样品的生药量为标准,换算成一致的量,计算各特征性成分的含量。通过SPSS 27.0软件,对数据进行单因子方差分析和显著性检验[24],结果见表4。 对含量测定结果进行系统分析。黄连-苦参比例为4∶1时,所得非洲防己碱、表小檗碱、巴马汀、小檗碱含量为各比例最高,且黄连总生物碱含量最高,与单药材提取具有显著性差异(P<0.05);黄连-苦参比例为5∶1时,所得苦参碱、槐定碱、木兰花碱含量为各比例最高,与单药材提取具有显著性差异(P<0.05);黄连-苦参比例为1∶3时,氧化苦参碱含量为各比例最高,与单药材提取具有显著性差异(P<0.05);黄连-苦参比例为1∶1时,苦参总生物碱含量为各比例最高。与黄连、苦参各药材单提相比,各比例下苦参中总生物碱类成分的溶出均有不同程度的提升,黄连中总生物碱类成分在黄连-苦参5∶1及4∶1比例下溶出表现为提升,其他比例表现为降低。随着药对中黄连比例的降低,黄连中整体生物碱类成分呈现下降趋势。对苦参中差异性成分进行比较,随着药对中黄连比例的降低,苦参碱、槐定碱在药液中的溶出降低,而氧化苦参碱的溶出提升,3种成分呈现“U”型分布,提示3者之间的相互影响关系。 3 讨论 本研究考虑与临床应用一致,黄连-苦参药对选择水回流提取法,选择分离效果最佳的乙腈-磷酸二氢钾溶液体系,对黄连及黄连-苦参药对的色谱条件进行优化,并在190~440 nm进行全波长扫描,于220 nm下进行指纹图谱建立以求全面对待测样品的差异性成分进行测定。结果表明,本研究建立的黄连-苦参药对指纹图谱稳定有效,可全面的测定黄连-苦参药对中的标志性成分。 大量文献研究发现,黄连-苦参药对在方剂中多采用1∶5至5∶1区间配比,故选择典型的9个配比进行量-质传递对比性研究。生物碱类成分作为黄连-苦参药对的主要药效成分,研究生物碱类成分在传统方剂煎煮过程中的溶出差异,可以为临床用药提供参考。故采用建立指纹图谱方式进行定性验证,确定稳定可测的生物碱类成分,并根据“2.1.8”项下结果,选择苦参中苦参碱、槐定碱、氧化苦参碱及黄连中木兰花碱、非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱进行研究[25-26]。 本研究在最佳吸收波长下,对黄连-苦参药对不同配比中10个差异性成分进行含量测定,分析差异性成分在不同配比下的溶出变化。苦参中3种差异性成分的溶出量随黄连比例的降低呈现“U”型分布,而黄连中7种差异性成分溶出量随黄连比例的降低整体呈现降低趋势。在黄连-苦参药对中,高黄连比例更容易促进药对中差异性成分的溶出。初步分析,当黄连-苦参药对中黄连占比的降低,可能会通过改变溶液中pH值、酸碱度等性质,对二者差异性成分的溶出产生影响,也可能对其中成分的相互转化产生促进作用,其具体产生机制有待深入研究。黄连-苦参药对被应用与各类中医经典方及现代经验方剂中[27-28],但其配伍面对临床不同疾病的合理应用仍需深入研究。 本研究首次将黄连-苦参相须药对与中医传统经验方剂药效相结合,探究差异性成分药理作用与临床疾病治疗的联系。黄连-苦参比例为5∶1时,所得苦参碱、槐定碱、木兰花碱含量为各比例最高;非洲防己碱、表小檗碱、药根碱、黄连碱、巴马汀、小檗碱含量较高,相比各药材单提含量有所提升,与单药材提取均具有显著性差异(P<0.05),与《普济方》中“相须为用,其效益彰”的方解一致,发挥各成分共同药效,达到“清热燥湿”效果。氧化苦参碱具有抗肿瘤作用,当黄连-苦参比例为1∶3时,其溶出量达到最大并与单药材提取具有显著性差异(P<0.05),与临床上使用参白解毒方进行抗结直肠道腺瘤[29]的治疗方式一致。药根碱可发挥降糖作用,在黄连-苦参比例为1∶1时含量最高,与国医大师李玉奇治疗消渴症时采用方剂中黄连-苦参药对[30]的配比一致,证明了方剂中黄连-苦参使用该比例配比的合理性。 综上所述,本研究成果预期可为开展黄连-苦参药对的量-效关系研究提供数据支撑,为临床不同疾病采用药对适宜配比用量、开发黄连-苦参药对新方剂提供借鉴。






 留言咨询
留言咨询
 留言咨询
留言咨询
 留言咨询
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP