推荐厂家
暂无
暂无
 留言咨询
留言咨询
 银牌4年
银牌4年
 400-860-5168转4383
留言咨询
留言咨询
400-860-5168转4383
留言咨询
留言咨询
 400-803-2799
留言咨询
400-803-2799
留言咨询
 400-875-7187
留言咨询
400-875-7187
留言咨询
 400-875-7187
留言咨询
400-875-7187
留言咨询




[align=center][b]如何避免液相色谱柱的损坏呢?[/b][/align]在高效液相色谱的使用中,我们会发现很多实验室会面临色谱柱频繁损坏的问题。[b]损坏的来源[/b]首先,需要来了解一下色谱柱的损坏来源于哪些方面:1、泵头密封圈的磨损2、进样阀转子密封圈的磨损3、析出的缓冲盐4、样品中的复杂易吸附基质其实,在工作过程中大多数的颗粒物堵塞导致的色谱柱背压升高问题是体现在柱头堵塞。而样品的易吸附机制也可能会和柱前端的硅胶表面游离的硅醇基发生作用导致色谱柱背压升高。但是这种情况在较低的PH系统中会有所改善,因为较低的PH系统会降低硅胶表面的硅醇基发生解离。[b]预防措施[/b]为了降低色谱柱的损伤问题,我建议大家在使用色谱柱时可以做一些预防。例如:[b]1在液相系统中加入在线过滤器[/b]在线过滤器的内部颗粒空隙一般是0.5微米,对于UHPLC会选择0.2微米空隙的在线过滤器。这个空隙尺寸和柱头的过滤筛板尺寸接近或者略小。所以,在线过滤器能够有效的阻隔来自样品、泵头密封垫、自动进样器的转子密封垫的颗粒物,以防止柱头过滤筛板的堵塞。注意:但是对于非常低死体积的液相系统,在线过滤器的加入可能会导致系统死体积增大峰展宽变严重的问题。而对于大多数的液相系统而言,色谱图不会有太大变化。[b]2在柱子前端使用保护柱[/b]保护柱有很多种,通用型、一体式、卡套式,但是它其实是一个和分析柱固定相相同的10-20mm的小柱子。因为它具有和分析柱相同的内部填料,所以保护柱不仅可以阻隔颗粒物,还可以阻隔易吸附的样品基质组分。注意:由于保护柱中可能有大量的强保留组分,在用强溶剂清洗系统时请取下保护柱,以防止这些强保留组分被洗脱进分析柱。保护柱如果出现吸附饱和也可能会对分离效果有影响,[b]3对样品进行净化处理[/b]有一些液相的分析方法没有明确标注样品的净化,但我希望大家在做分析时都能够考虑样品的净化,常用的方案是对样品进行过滤。[b]色谱柱的储存[/b]在实际工作中,色谱柱的储存也会影响柱子的使用寿命。那么对于色谱柱的储存,也有以下建议:[b]短期储存时[/b]应该保存在最接近常用的流动相条件下。并且,一定要移除系统中的缓冲盐、酸等。[b]长期储存时[/b]将色谱柱按照说明书的要求保存在对应的溶剂中,并且将色谱柱从系统中取下两头用堵头堵死。本文来源于微信公众号《色谱学堂》,本文对内容进行了部分修改
在进行[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析时,确保样品的稳定性以及避免维生素C的降解非常重要。以下是一些具体的方法和注意事项: 样品采集与处理:样品采集后要尽快进行处理和分析,以减少维生素C的暴露时间和降解机会。在处理过程中,要轻柔操作,避免剧烈搅拌或高温处理,这些都可能加速维生素C的降解。 使用合适的保存条件:将样品存放在低温环境中,如冰箱(4°C),可以显著减缓维生素C的降解速率。另外,避免样品暴露在阳光下或强光下,因为光照也会加速维生素C的分解。 添加稳定剂:在样品中加入适量的稳定剂,如抗坏血酸(维生素C本身)或其他抗氧化剂,可以帮助保护维生素C不被氧化。此外,使用一些酸性缓冲液(如磷酸盐缓冲液)调节样品的pH值至酸性范围(pH 2-4),也能有效抑制维生素C的降解。 选择合适的样品瓶和密封盖:使用高质量的样品瓶和密封盖,确保样品的密封性,防止空气和水分的侵入,从而避免维生素C的氧化降解。优选使用具有良好密封性能的聚丙烯或氟塑料材质的样品瓶。 控制进样量:在[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析过程中,要精确控制进样量,避免进样过多或过少影响分析结果的准确性。同时,尽量减少样品在进样器中的停留时间,以减少维生素C的降解。 定期校准和维护仪器:定期对[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]进行校准和维护,确保仪器的稳定性和准确性。在分析过程中,要保持仪器的清洁和良好的工作状态,避免因仪器问题导致的分析误差。 总之,在进行[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析时,要综合考虑样品的采集、处理、保存、进样以及仪器维护等多个环节,采取有效的措施确保样品的稳定性,避免维生素C的降解,从而获得准确可靠的分析结果。
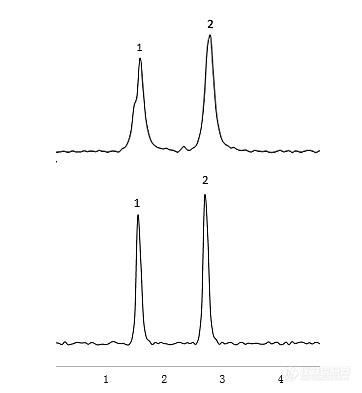
[b][font=宋体][back=white]问题描述:“溶剂效应”会导致哪些异常峰型?如何避免溶剂效应?在[/back][/font][back=white]HPLC[/back][font=宋体][back=white]分析中样品溶剂的选择与流动相有什么关系?[/back][/font][font=宋体][back=white]解答:[/back][/font][/b][font=宋体][back=white]([/back][/font][back=white]1[/back][font=宋体][back=white])当样品溶液的溶剂强度强于流动相溶剂强度时可能会导致峰前端展宽、峰分叉,即色谱图上较早洗脱的峰扭曲变形或者开叉,与此同时较晚洗脱的峰则较为尖锐与对称,这种现象一般称之为“溶剂效应”。[/back][/font][font=宋体][back=white]([/back][/font][back=white]2[/back][font=宋体][back=white])这里的强溶剂可以理解为洗脱能力强的溶剂,色谱常用的有机相是甲醇与乙腈,纯乙腈就是洗脱能力最强的溶剂。流动相的洗脱能力越强,在其他条件不变的情况下,出峰越快,保留时间越短。[/back][/font][font=宋体][back=white]([/back][/font][back=white]3[/back][font=宋体][back=white])液相色谱分析中产生“溶剂效应”的原因主要有:[/back][/font][back=white]a.[/back][font=宋体][back=white]样品溶液的溶剂强于流动相。如图[/back][/font][back=white]5-5[/back][font=宋体][back=white]所示,当样品溶液的溶剂是[/back][/font][back=white]100%[/back][font=宋体][back=white]乙腈(强溶剂),而流动相组成较弱([/back][/font][back=white]18%[/back][font=宋体][back=white]乙腈[/back][/font][back=white]/72%[/back][font=宋体][back=white]水):第一个峰是开叉的,并且与第二个峰相比,明显地变宽了(图[/back][/font][back=white]5-5[/back][font=宋体][back=white]上);当样品溶液的溶剂变成流动相时,所有的峰形都改善了,且变得尖锐(图[/back][/font][back=white]5-5[/back][font=宋体][back=white]下)。这是因为当样品进样时,有可能出现峰展宽,最佳的样品溶液组成和体积将会保持在[/back][/font][back=white]10%[/back][font=宋体][back=white]甚至更低,在这个例子里,当样品溶剂与流动相溶剂强度不同时,此时有些样品分子溶解在强溶剂中,并随强溶剂流过柱子,而有些则溶解在流动相中,从而导致峰分叉。[/back][/font][align=center][img=,362,393]https://ng1.17img.cn/bbsfiles/images/2021/03/202103221545160685_5875_3389662_3.jpg!w362x393.jpg[/img][/align][align=center][i][font=宋体]图[/font]5-5 [font=宋体]样品溶剂对组分峰型影响[/font][/i][/align][back=white]b.[/back][font=宋体][back=white]进样量比较大。[/back][/font][back=white]c.[/back][font=宋体][back=white]样品的[/back][/font][back=white]pH[/back][font=宋体][back=white]值与液相色谱流动相[/back][/font][back=white]pH[/back][font=宋体][back=white]悬殊,比如酸性流动相体系,样品[/back][/font][back=white]pH[/back][font=宋体][back=white]偏碱性([/back][/font][back=white]pH=10[/back][font=宋体][back=white]),这种情况下也很有可能发生“溶剂效应”。[/back][/font][font=宋体][back=white]([/back][/font][back=white]4[/back][font=宋体][back=white])其实上面三个原因归结起来就是一个原因:样品的溶剂与流动相存在一些不一样。当溶解样品的溶剂不同于流动相时,样品溶剂与流动相发生混合,样品溶剂被冲稀。如果进样溶剂之强度高于流动相,样品在瞬间会表现为在较强溶剂中,并以较快速度通过色谱柱,表现在色谱图上就是:色谱峰的保留时间缩短。当进样溶剂与流动相混合时,一部分分子会先与流动相混合,致使这些分子通过色谱柱的速度发生变化,使峰形扭曲,发生变形。[/back][/font][font=宋体][back=white]([/back][/font][back=white]5[/back][font=宋体][back=white])所以要解决这个问题的最佳方案就是,使用流动相的起始梯度溶解样品,如果由于溶解度的问题,不得不改用其余的溶剂,那就只能降低进样体积来解决。[/back][/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]










 我要推广仪器
我要推广仪器
 下载APP
下载APP




