

风之彩
第1楼2009/04/04
3 色谱条件的建立
这是过程可能很短很简单,也可能很漫长很费事,关键和很多因素有关,如化合物的性质、实验室现有的条件、成本、检测对灵敏度的要求等等,总之,就是要在满足检测要求的前提下,分析测试时间越短、成本越低、操作越简单越好。
1)检测波长的选择
文献报道的检测波长有225nm和275nm。一般说来(也有化合物例外),化合物在低波长段信号响应会好些,但是干扰也多,随着波长的增加,干扰会逐渐降低。不确定这两种波长的响应究竟差多少,我就拿紫外扫描了一下,的确225nm左右有最大吸收,而275nm处吸收太低,不可能满足我检测的要求,所以选择225nm作为我的检测波长。
很希望275nm处有强的紫外吸收,这样就算比225nm处稍微响应低些,但是干扰少,基线平,色谱条件建立起来容易啊,可惜……
2)色谱柱、流动相和内标的初步选择
选择色谱柱主要是参考文献和实验室现有色谱柱的情况, C18柱(ODS柱)适合大多数非极性样品的分析,如果样品极性大,可以选择氨基柱、腈基柱和HILIC柱等,也可以选择正相系统。只是从反相系统向正相系统过渡的时候要注意,不然会引起整个系统的堵塞。
我需要测定的物质A极性不大,文献报道基本都是C18柱,所以就用我们实验室最常用、储备也比较多的几根柱子开始摸条件,如Waters Symmetry C18,Agilent ZORBAX SB C18等(当然也是考虑了流动相的性质)。
流动相考虑到成本和毒性,决定先试甲醇,水相就参考文献配了25mmol/L的KH2PO4溶液(pH3.0)。
内标先试用了实验室现有的化合物,但是均与A的保留时间差的比较大,且相对靠前(血浆样品容易有干扰)。后来选用了文献报道的化合物B,出峰在A的后面,干扰少。
总共比较了Waters Symmetry C18(4.6×150mm, 5μm) 、Waters Symmetry C18(4.6×75mm, 3.5μm)和Agilent ZORBAX SB C18(4.6×150mm, 5μm)三根柱子,最后选择了第二根色谱柱(尽管没有文献报道是用这么短的柱子的),原因是:
1. Waters的这两根柱子都是Symmetry C18的,差别是长度和粒径,短柱子样品出峰快,样品与内标分离效果好,但是就是柱压高些。
当用150mm的柱子进样,甲醇:水相=72:28(v:v),1ml/min, 225nm, 柱温35℃时,样品A的保留时间是tR=8.2min,内标B的保留时间tR=14min左右。这样的进样时间对于我来说太长了, 2300多个样品,保留时间的长短直接影响到我的进样时间,我希望保留时间再短些,这就意味着有机相的比例还要增加,但是由于水相有盐,有机相太高怕析出,而且成本也会增加,所以就选择了短柱。
2. Agilent的柱子样品和内标的分离效果没有远没有Waters的好,如果要分开,那么两者的保留时间又会大于10min,但是柱压明显比Waters的低。
风之彩
第2楼2009/04/04
3)色谱条件的优化及前处理方法的选择
我的原则就是前处理越简单越好,分析时间越短越好,分析成本越低越好。
1.先拿样品及内标的对照品来初步确定色谱条件
当然这只是初步看一下,看看标准品进样,灵敏度怎样,是否需要浓缩样品,内标和样品峰分的怎么样等等。
2.根据检测要求选择最方便的前处理方法
根据我的检测要求,LLOQ最少要做到500ng/mL,当然能低更好。看看我的灵敏度,用蛋白沉淀法应该没有问题,当然前提是没有内源性干扰。于是试了几种最常用的沉淀剂:甲醇、乙腈和甲醇硫酸锌。
一般三者加的量是甲醇硫酸锌最少、乙腈次之、甲醇最多,当然是为了保证蛋白能充分沉淀。加的沉淀剂越少,样品稀释就越少,可能就会有更高的灵敏度。
结果,硫酸锌沉淀响应最好,峰也窄,而甲醇和乙腈沉淀样品峰响应相差不大,但乙腈沉淀峰更宽些,因此先选择用甲醇硫酸锌沉淀。
三种沉淀剂比较(其中硫酸锌沉淀样品的进样量为其余两种的1/2),1是样品峰,2是内标峰。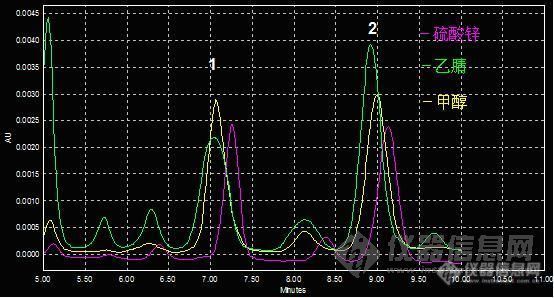
风之彩
第3楼2009/04/04
3.优化色谱条件
确定好了色谱柱、流动相、内标和前处理方法,下面就是要正式开始优化条件了,有干扰的避干扰,能缩短分析时间就缩短分析时间。
1)首先调节流动相比例、柱温,使得样品峰和内标能够分开,且总的分析时间在10min以内。
2)做空白血浆加标的样品,看有无干扰。果然,不出所料,样品峰和内标处均有干扰,继续优化流动相比例、柱温、流速等参数,样品峰处基本无干扰了,但是干扰峰始终在内标峰出徘徊。
3)下面开始了排除血浆干扰的优化工作。
A.走梯度:甲醇走梯度,基线波动太大,有机相换成乙腈,样品峰响应下降,峰变宽,也不合适,失败。
甲醇走梯度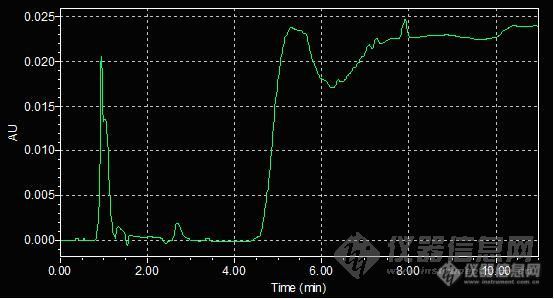
有机相改成乙腈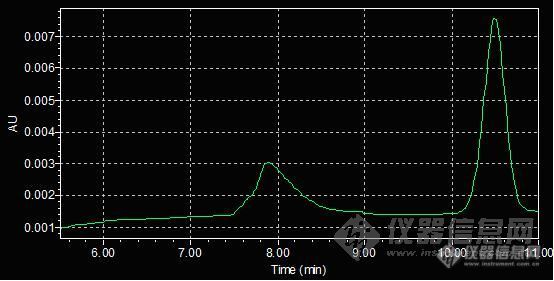
B.前处理改用提取:虽然不愿意,但是用乙醚还是试了一下,结果,提取还是有干扰,也不行。
C.改变流动相水相的pH值:调了几个pH值 3.0, 3.6, 4.3,结果发现,干扰峰对于流动相的pH值比较敏感,随着pH值的升高,干扰峰的保留时间会降低,当然样品峰和内标峰也会有同样的趋势,只是没有干扰峰那么敏感。最后把pH值调成3.4时,干扰峰终于可以在样品峰和内标峰中间出峰。而且做了6份不同来源的血浆,干扰峰均在样品峰和内标峰中间出峰。
pH 4.3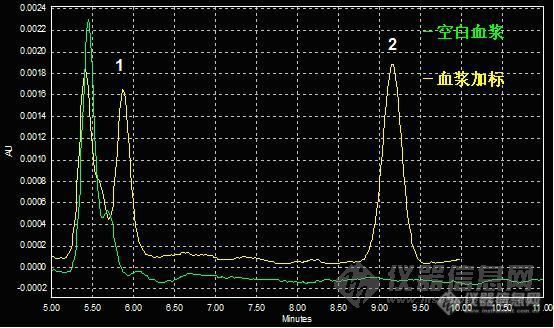
pH3.6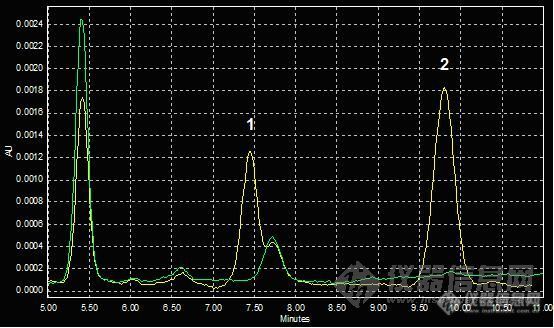
pH3.0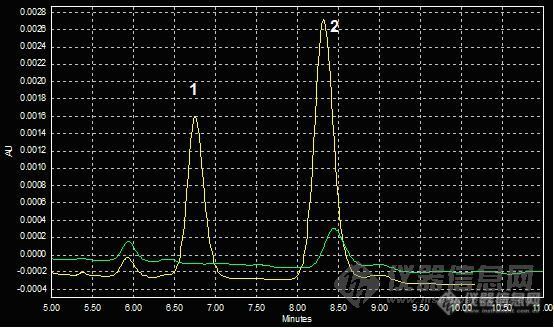
pH3.4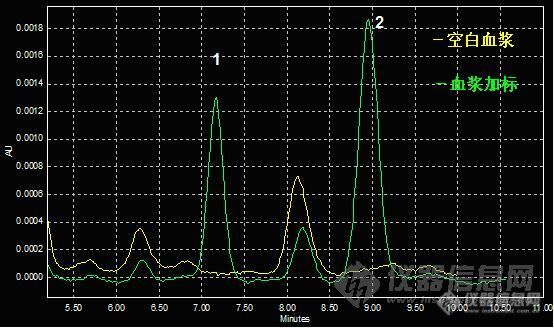
4)但是新的问题出来了,样品峰和内标峰的保留时间出现了漂移,老是不稳定,怀疑可能缓冲液的缓冲能力不够,于是缓冲盐的浓度加大了一倍,果然,保留时间稳住了,呵呵!
5)下面开始配血浆标准曲线,看线性。线性还是很好的,每次都能达到3个9以上,但是柱压阿,一直让我很担心。进一针,往上跑一点,虽然每次冲柱后能降一些,但是还是有上升的趋势。这对于我大批量样品而言,是很不合适的。看来用甲醇硫酸锌沉淀不太合适。尽管以前这种沉淀剂我也用过,但是那时候是Agilent的柱子,且粒径是5μm的。可能对于Waters的这一型号的柱子,用盐或者是不太完全的蛋白沉淀方法不太合适。
6)有得重新选择沉淀剂,最常用的还是甲醇和乙腈。乙腈沉淀效果好,所需的用量也相对少。但是样品在乙腈中响应就是低,峰也太宽。不行还是得用甲醇。可是问题来了,甲醇的用量大,样品的稀释倍数就多。不过甲醇沉淀,也基本能满足我500ng/mL的LLOQ的要求。但是峰形就是没有我用硫酸锌沉淀的好。
7)尝试优化峰形。考虑到甲醇硫酸锌沉淀中是有水(用来溶解硫酸锌的),可能是受到溶剂效应的影响,我就尝试着在甲醇沉淀后,上清液又加水的方法,果然,峰形变好看了,峰也瘦了,而且虽然加了水,但是峰高与不加水比基本没有下降。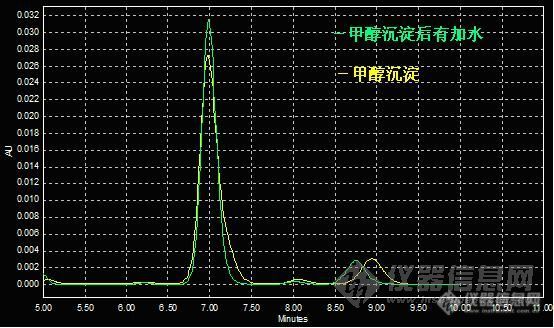
8)最后我用30μL进样,LLOQ能做到200ng/mL。
风之彩
第4楼2009/04/04
4.测定预试样品,新问题出现
预试测定了100个血浆样品,检测限没有问题,但是新的问题还是出现了,发现我的标准曲线样品的干扰峰是在样品和内标中间的,但是4个受试者的血浆干扰峰均在内标峰出峰的地方。
我做标准曲线的血浆购自血站,是病毒灭活过的。但是受试者的血浆,是抽的招募健康受试者的血直接离心得到的。难道是这个差别。然后我试了实验室其他的从健康受试者身上采的的血浆做了验证,果然,干扰峰均在内标出峰的地方出峰。
这是我用买来血浆做的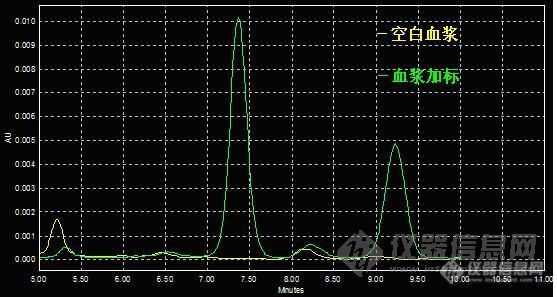
这是受试者的血浆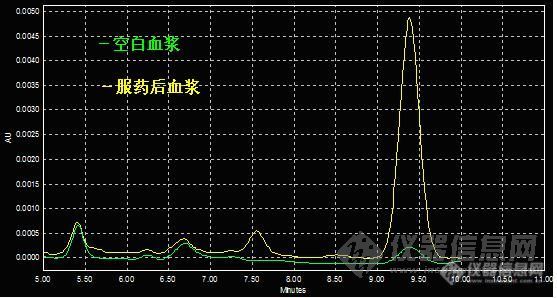
5.重新优化色谱条件
翻阅了实验记录,还是决定先调节水相的pH值来试试,是往酸还是往碱调?发现干扰峰基本在内标快出完的时候出,所以决定还是往酸调,把它往后推。就调回了3.0,果然,两者分开了,基线分离。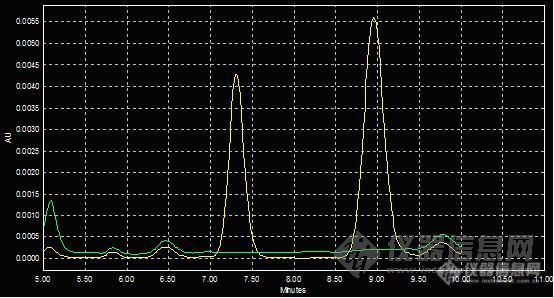
并且我又可以把缓冲盐的浓度降下来了,仪器因为高盐会析出而损坏等的风险也可以降低些了。
到此,我的方法优化正式结束,加上预试后重新优化,经历了2个月的时间。现在方法学考查也已经完成。
风之彩
第5楼2009/04/04
最后,做个小结,也算对用HPLC做生物样品分析(血浆、尿、组织等)的版友介绍一下我的个人体会,供大家参考:
1 首先就是要查阅相关资料,如文献等,了解你要测化合物的性质,根据文献来选择你的初始条件。比如色谱柱、流动相、前处理方法,还有检测器等等。
2 了解液相的基本原理和基本操作,这是你做好液相、用好液相的前提。比如什么样品可以用反相、什么样品用正相系统来检测,反相系统加大有机相的比例峰会怎么变化;柱温、流动相pH值、溶剂对你的样品峰会有什么影响;遇到一些简单的仪器问题怎么处理、一些仪器参数是什么意思、对你的样品峰有什么影响等等,当然这些是慢慢学习的过程,学的越多,你会发现你建立方法用的时间越短、走的弯路越少。当然,我也在慢慢的学习中。
3 知道各种前处理方法及其优缺点,比如沉淀剂的种类、加入的比例,适合的化合物性质。常用的提取溶剂有哪些,适合提取的化合物的极性等。
4 要预先估计你所需要的LLOQ,根据这个LLOQ来优化你的色谱条件。比如灵敏度足够,可以用沉淀,不够可以用提取,提取浓缩的比例根据LLOQ来定。当然还可以其他的辅助手段,比如加大进样量、用梯度洗脱的方式等等。
5 做液相可能最头疼的就是干扰,用紫外检测器特别是在低的检测波长段,可以说是杂草丛生,所以一般分析时间都比较长。改用提取后,可能会好些。如果可以,宁愿牺牲一些灵敏度而选择相对高一些的检测波长。荧光检测,干扰就很少。多试不同来源的空白基质,确保没有干扰。有条件,也像我这样,买来的空白血浆和受试者的空白最好也试一下,以免不同(这样的情况非常非常少见)。
先写这么多,比较乱,有空再来上点图谱。其中有写的不当的地方,请大家批评指正。

































