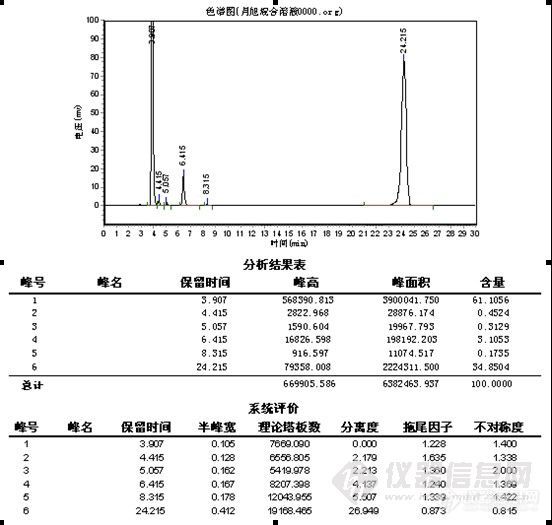
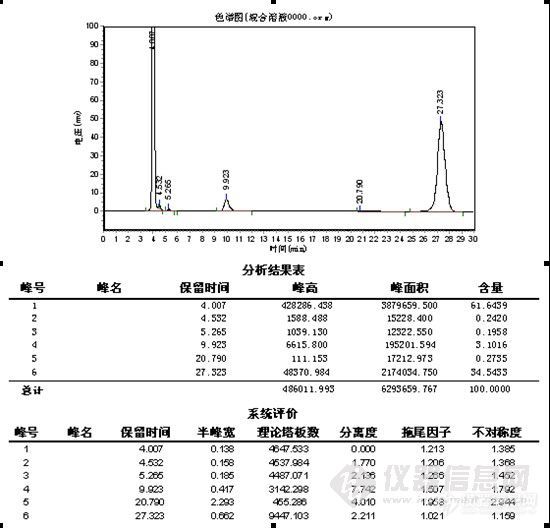
zxhcnf
第2楼2009/11/04
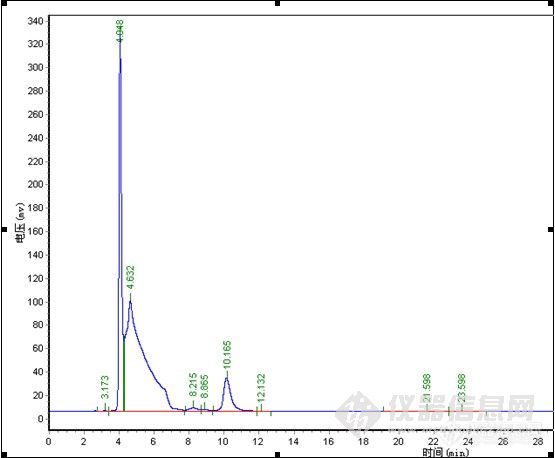
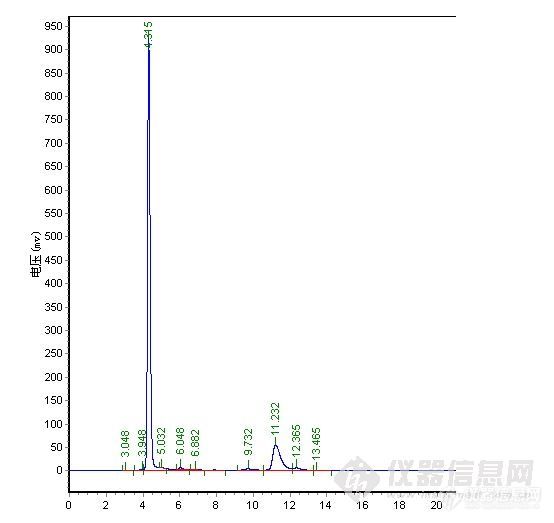
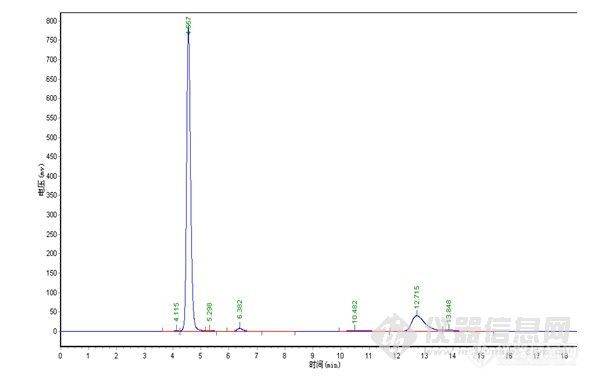
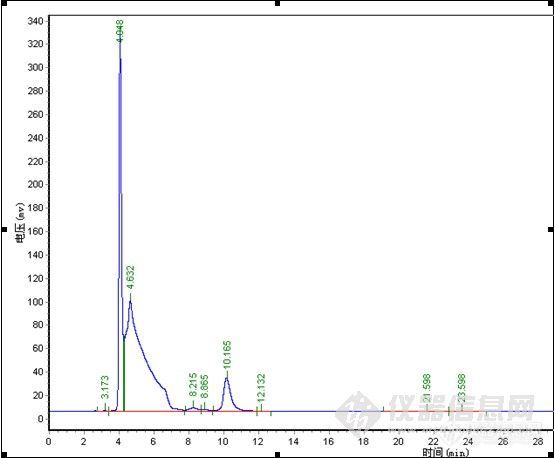
楼主真是费了不少心思,峰形出现异常找原因很麻烦,象碰到容易分解的样品查起原因来更是不好弄,有时候一看见怪峰慌了神,赶紧查液相和柱子的问题呵呵。。。其实做方法开发就象楼主这样先是要对这个样品的性能有一定的了解,样品是否稳定,在甲醇、水或乙腈中的溶解性,还有分子结构等,这些都对成功开发方法具有重要的作用。楼主很快的查明了原因,做事真是细心。谱图上有点问题我觉得有些还可以改善一下,1)水解产物C的峰在4min左右,出峰时间太快,靠近死体积出峰,这个区域会有比较多的杂质干扰,所以我认为增大流动相中水相的比例,让C峰的出峰时间延长一点可能会更好;2)原料A的峰,峰形有点前延,可以有改善的空间,不过它不是目标分析物,可以少放点心思在这上面,如果认为需要改善,这个帖子可能对你有点用。http://www.instrument.com.cn/bbs/shtml/20091030/2184029/