wanttofly
第2楼2010/12/14
二.红外特征知识
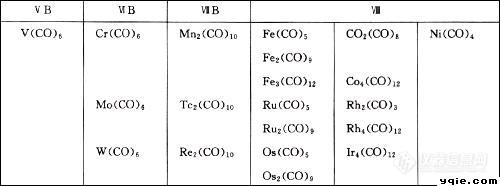
随着各种金属羰化物不断被合成,其结构和光谱特征也做了大量的研究,该方面的著作和文献也很多,在一些红外的专业书籍中也将其作为一个分支加以表述。下面的一些论述主要引用了下面2本书的论述:
[1] 中本一雄著.无机和配位化合物的红外和拉曼光谱.黄德如,汪仁庆译.北京:化学工业出版社,1991.
[2] 吴瑾光主编.近代傅里叶变换红外光谱技术及应用.北京:科学技术文献出版社,1994.
大多数羰基配位化合物约在2100—1800cm-1处出现强而尖锐的νco带。通常,该谱带不与其它的振动模式偶合,也不会出其它振动谱带的出现而被掩盖。所以,仅仅研究该谱带,常常便可以得到有关羰基配位化合物的结构和成键的有价值的信息。与游离的C0的νco(2155cm-1)相比,大多数羰基配位化合物的νco向低频方向位移。
简单的分子轨道理论可对这一现象解释如下:首先,CO向金属原子的空轨道提供5σ电子形成σ键。由于5σ轨道是弱反键轨道,因此,σ键的形成有助于提高νco。其次,金属原子又反馈给dπ电子给CO的空反键轨道2Pπ*形成π键,这有助于降低νco。这两个成键分量是协同作用的。当金属处于较低的氧化态时,协同作用的净结果是电子从金属向CO迁移,因而,金属羰基配位化合物的νco一般比游离的CO的来得低。
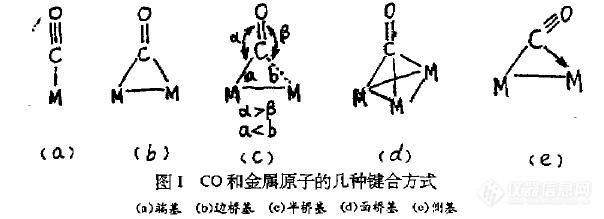
端基配位形成M-C-O直线形结构,其νco一般在2125-1900 cm-1处,而在边桥基配位方式中,CO同时和两个金属原子键合,其νco位于在1900-1750cm-1范围内,如[Pt6(CO)12]红外谱图中,1990cm-1处谱带对应于端基,而1818cm-1和1795cm-1处对应于边桥基。因此在实际应用中,用红外光谱区分端基和边桥基两种配位方式是十分方便。
当含有其它配体时,νco发生变化。当CO被卤原子取代后形成金属羰基卤化物,νco将向高频方向位移,因为形成M-X键后,金属的电正性增强了,M→CO的反馈键减弱了。在一系列同类型卤代羰基化合物中,氯代物的νco最高,碘代物的νco最低,溴代物的则位于两者之间,正如从它们的电负性所预期的那样。
当CO被受电子能力弱的配体取代,νco减小。例如当L为亚膦酸酯配体,νco按照Ni(CO)3L、Ni(CO)2L2、Ni(CO)L3顺序逐渐减小,这是由于L有较强的电子给予能力和较弱的电子接受能力,使得配位化合物中M→CO的反馈键加强,从而使νco减小,随着L数目的增加,M→CO的反馈键越强,而νco趋于减小。
备注:看起来挺罗嗦的,我简要总结就是:金属羰化物的羰基伸缩振动在2100—1800cm-1,这个区域吸收比较少,有实用价值,可以用于区分端基和边桥基。另外金属羰化物还会连上配体,羰基吸收会发生变化,具体数据有很多书和资料来查阅。
wanttofly
第4楼2010/12/14
三.金属羰化物红外光谱的应用
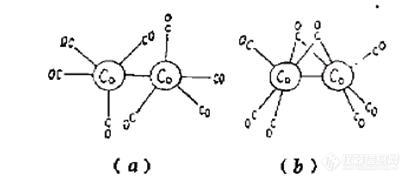
3.1 催化剂合成表征:这个是最简单的应用,就是合成了金属羰化物,然后看看是否正确,只要对一下文献的数据就可以了。当然由于金属羰化物性质不稳定,构型也会发生变化,所以也会出点意外(比如后文的八羰基二钴),不过这些纯物质前人已经研究够多了,多看看资料就行了。
3.2 反应机理推断:这个是搞合成研究的人写文章的重点内容,如果你看得类似的文章多了,会有点糊涂,不仅数据不一样,解释也可能不一样,所以我也不太明白那些研究人员是如何能够在复杂的体系中得出结论的。有些研究人员看了文章也让我来测,但是我没有原位池,无法跟踪中间过程,而金属羰化物在空气中构型是会变化的,因此取出的反应液中间谱图做了,但结果不知道是不是能够让他们得到合理的解释。
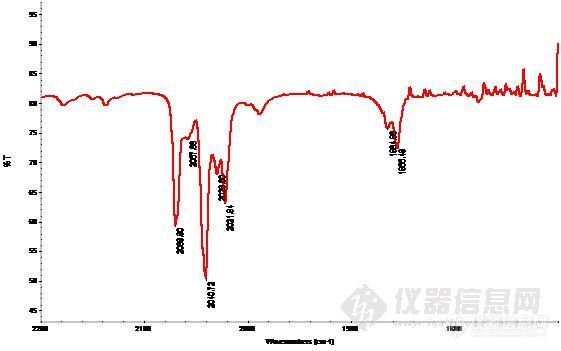
例:在别人论文里摘抄的:[RhI2(CO)2]-的羰基伸缩振动频率位于2064cm-1和1989cm-1处,用CH3I处理该化合物,红外吸收出现在2062cm-1和1711cm-1,后者被认为酰基络合态;加入CO产生一个新物种,其红外谱带位于2141、2084和1708cm-1,该物质缓慢分解为[RhI2(CO)2]-,故被指认为[RhI3(COCH3)(CO)2]-。我测的类似体系:1700cm-1左右的吸收被反应液中的羰基覆盖的,但是2000cm-1还是能看出来的有那么两个峰,但是具体位置用它的理论还不好解释。
3.3 反应控制和表征:这实际是应用2的衍生,通过金属羰基红外吸收的变化来推断催化剂的活性。人家跟我说有篇专利用红外来测定一价铑和三价铑的比例,我直接说不可能,后来拿来资料,知道点红外知识的也知道它所说的峰其实就是和不同价态的铑连接的羰基的伸缩振动。