

离子色谱标准加入法测凝析水中的硫酸根
adongsun
第4楼2011/08/21
这个论坛的快速回复居然不能复制的,晕倒,刚写了几百字的回复没了。
再来一遍吧。
技术贴要支持的!
首先,你的噪音0.0077如果单位是us/cm,那确实大了点,通常是要再小一个数量级的。但我跟你的想法一样,这不是影响你结果准确度的关键。
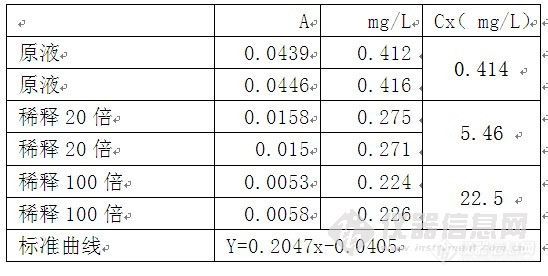
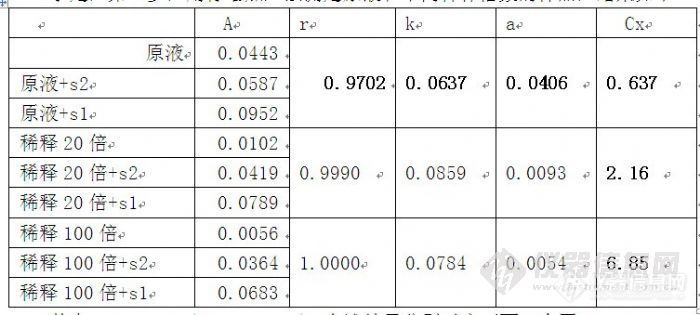
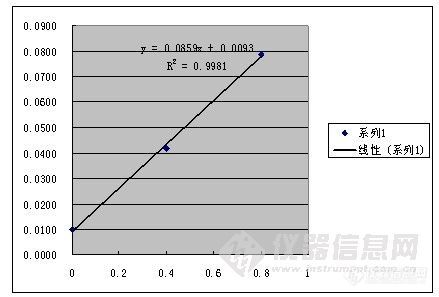
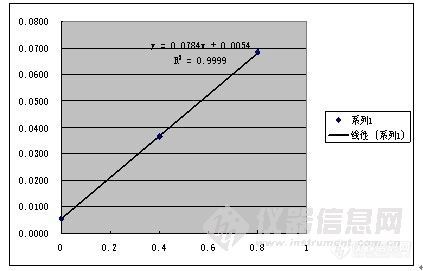
其次,标准加入法通常应用于分离有干扰的基体,不知道你的硫酸根附近有啥分不开的物质。或者你稀释用的纯水不干净?标准加入法2个点回归,好像少了点,如果追求准确度的话,我觉得3个点是至少的。
最后,是否稀释理论上应该不影响结果的准确度,但是跟你分析的一样如果线性范围有问题,那准确度一定会丧失。所以你要提供你校正用的线性范围,这样大家可以帮你再进一步分析。并且需要告知是碳酸体系还是氢氧根体系,这对相关度是否是线性也有关。

































