=======================================================================
【活动内容】

PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。
下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。
999youran
第3楼2017/08/03
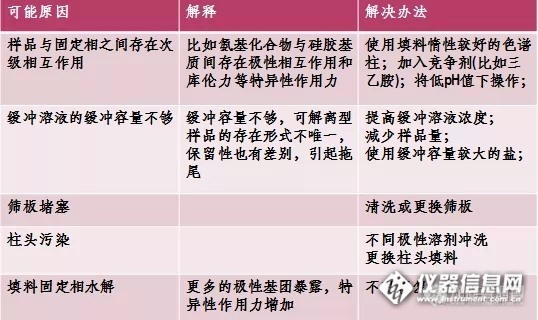
气相分析中峰拖尾的原因是复杂多样的,如果遇到峰拖尾,不妨按照以下分类情况进行排查:?
1. 只有活性组分拖尾活性组分指的是极性较强或热稳定性低的化合物,样品流路中的活性位点最容易导致活性组分的吸附和分解,可以重点检查:
? 提高样品流路的洁净度,重点对进样口和色谱柱进行维护
? 提高样品流路的惰性,尽量选用惰性可靠的产品,如超高惰性样品流路配件-- 超高惰性衬管、分流平板、色谱柱等
? 正确切割和安装毛细管柱,色谱柱的切割面应平整光洁,残留的毛边或碎屑都会是潜在的活性位点;注意色谱柱在FID/NPD/FPD等检测器喷嘴内探伸的距离不宜过短,因活性组分有可能被喷嘴的金属管壁吸附而拖尾
2. 挥发性组分拖尾
早流出组分拖尾严重。多在不分流进样、柱上进样或样品溶剂与色谱柱极性不匹配时出现,改善峰形的方法可以是:
? 采用保留间隙柱
? 降低进样口温度50?C
? 调整程序升温初始温度于溶剂沸点10~25?C之下
? 确保色谱柱安装没有漏气,样品流路各连接处没有死体积
3. 低挥发性组分拖尾
拖尾峰多是较晚流出的色谱峰,拖尾随保留增加而加剧。
? 检查系统是否存在污染
? 消除样品流路中的冷凝点,适当提高各部位温度
? 减少系统死体积
4. 所有组分都拖尾
主要原因可能是:
? 样品流路严重污染
? 进样口总流量过低
? 色谱柱安装位置不当
5. 其他可能导致峰拖尾的原因包括:
? 不分流模式下,延迟时间过长
? 进样技术不佳或进样时注射器中有样品残留
? 检测器尾吹气流量不足
? PLOT色谱柱过载
? 组分共流出
? NPD检测有机磷选择了白色铷珠
yifan1117
第4楼2017/08/03
理想的色谱峰遵循正态分布,应该符合高斯曲线,是完美而对称的峰形(如图A);然而由于化合物性质的特殊性、色谱柱故障、操作条件等问题存在,峰形可能会不对称,前延(B)或拖尾(C),在此将引起峰形不理想的原因陈述一下。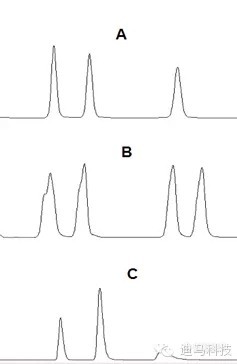
(1)峰形前延(B):
(2)峰形拖尾(C):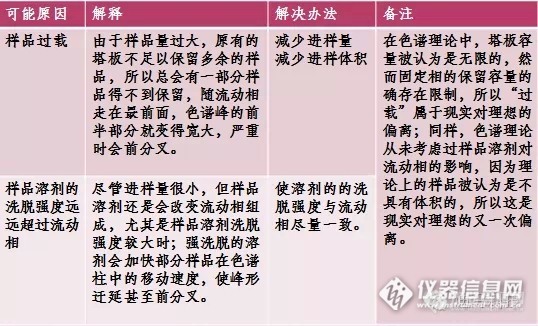
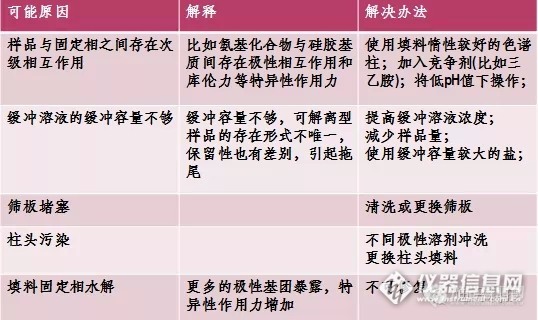
总结:
图谱前沿和拖尾的原因主要是流动相选择不合适,可以相应调整流动相的极性,或者适当加入酸来调整,可以得到较好的改善。一般来讲,酸碱在流动相中对于前沿和拖尾影响较大。
柱前沿是可能因为柱超载,拖尾是可能因为样品被污染,选择合适的流动相,调节好PH能够改善这以情况。
而产生托尾峰往往是有机性相近杂质没有分开,可以优化分析方法,或更换柱子试一试;也可能由于柱子使用时间太久柱效下降出现塌陷等原因;再有也会根样品本身性质有关,基团需要流动相中添加能与之结合优化峰形的化学物质,要根据具体情况而定。柱子可能污染了吧,溶剂也可能有,或柱效下降等等
sixingxing
第7楼2017/08/03
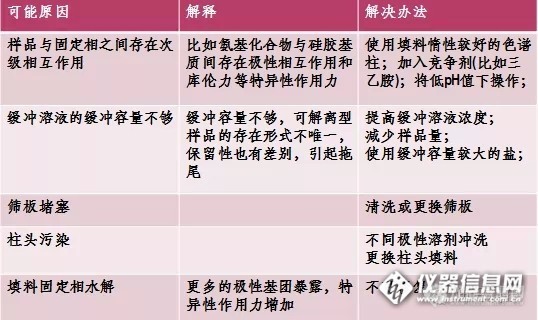
气相分析中峰拖尾的原因是复杂多样的,如果遇到峰拖尾,不妨按照以下分类情况进行排查:?
1. 只有活性组分拖尾活性组分指的是极性较强或热稳定性低的化合物,样品流路中的活性位点最容易导致活性组分的吸附和分解,可以重点检查:
? 提高样品流路的洁净度,重点对进样口和色谱柱进行维护
? 提高样品流路的惰性,尽量选用惰性可靠的产品,如超高惰性样品流路配件-- 超高惰性衬管、分流平板、色谱柱等
? 正确切割和安装毛细管柱,色谱柱的切割面应平整光洁,残留的毛边或碎屑都会是潜在的活性位点;注意色谱柱在FID/NPD/FPD等检测器喷嘴内探伸的距离不宜过短,因活性组分有可能被喷嘴的金属管壁吸附而拖尾
2. 挥发性组分拖尾
早流出组分拖尾严重。多在不分流进样、柱上进样或样品溶剂与色谱柱极性不匹配时出现,改善峰形的方法可以是:
? 采用保留间隙柱
? 降低进样口温度50?C
? 调整程序升温初始温度于溶剂沸点10~25?C之下
? 确保色谱柱安装没有漏气,样品流路各连接处没有死体积
3. 低挥发性组分拖尾
拖尾峰多是较晚流出的色谱峰,拖尾随保留增加而加剧。
? 检查系统是否存在污染
? 消除样品流路中的冷凝点,适当提高各部位温度
? 减少系统死体积
4. 所有组分都拖尾
主要原因可能是:
? 样品流路严重污染
? 进样口总流量过低
? 色谱柱安装位置不当
5. 其他可能导致峰拖尾的原因包括:
? 不分流模式下,延迟时间过长
? 进样技术不佳或进样时注射器中有样品残留
? 检测器尾吹气流量不足
? PLOT色谱柱过载
? 组分共流出
? NPD检测有机磷选择了白色铷珠
lijing320323
第8楼2017/08/03
出现前伸峰的原因:进样量或样品浓度高,溶解样品的溶剂较流动相极性强;保护柱或色谱柱污染或失效。
方法:①柱温低:升高柱温;②样品溶剂选择不恰当:使用流动相作为样品溶剂;③样品过载:降低样品含量;④色谱柱损坏:更换柱子。
拖尾峰:
柱超载,降低样品量;增加柱直径采用较高容量的固定相;峰干扰,对样品进行清洁过滤;调整流动相;硅羟基作用,加入三乙胺,用碱致钝化柱增加缓冲液或盐的浓度降低流动相pH值;柱内烧结不锈钢失效,更换烧结不锈钢;加在线过滤器,对样品进行过滤;死体积或柱外体积过大,将连接点降至最低;尽可能使用内径较细的连接管;柱效下降,更换柱子;采用保护柱,对柱子进行再生。