

植物源性食品中百草枯和敌草快残留量的测定
液相色谱-质谱/质谱法
1 范围
本方法规定了植物源性食品中百草枯和敌草快的制样和液相色谱-质谱/质谱测定方法。本方法适用于大米、大豆、玉米、小麦、棉籽、干木耳、甘蓝、苹果、香蕉、草莓中百草枯和敌草快残留量的测定和确证。质谱条件:参见附录A,农药方法物质的液相色谱-质谱/质谱图参见附录B,精密度见附录C。
2 原理
试样中百草枯和敌草快残留用甲醇-盐酸溶液匀浆提取,经弱酸性阳离子交换固相萃取柱净化后,用液相色谱-质谱/质谱仪测定,外标法定量。
3 试剂和材料
除另有规定外,所用试剂均为分析纯,水为GB/T 6682规定的一级水。
3.1 试剂
3.1.1 乙腈:色谱纯。
3.1.2甲醇:色谱纯。
3.1.3甲酸:色谱纯。
3.1.4 盐酸
3.1.5 氢氧化钠
3.1.6 盐酸溶液(0.1mol/L):移取9ml盐酸,用水定容至1L。
3.1.7 氢氧化钠溶液(1mol/L):称取4.0g氢氧化钠,用水溶解,并定容至100ml。
3.1.8 甲酸溶液(0.1%):移取1.0ml甲酸,用水稀释,并定容至1L。
3.1.9 乙腈-0.1%甲酸溶液(1+1,体积比):量取50ml乙腈,加入50ml0.1%甲酸溶液(3.1.8),混匀。
3.1.10 乙腈-水-甲酸溶液(88+10+2,体积比):量取88ml乙腈、10ml水和2ml甲酸,混匀。
3.1.11甲醇-0.1mol/L盐酸溶液(1+9,体积比):量取900ml0.1mol/L盐酸溶液(3.1.6),加入100ml甲醇,混匀。
3.2材料
3.2.1 Oasis WCX固相萃取(SPE)柱:60mg,3ml或性能相当者,使用前依次用1ml甲醇、1ml水活化。
3.2.2 微孔滤膜:0.22 um,有机系。
3.3 方法溶液
3.3.1 方法储备溶液
敌草快二溴盐方法物质:C12H12Br2N2,CAS号85-00-7,纯度大于或等于99.0%。
百草枯二氯盐方法物质:C12H14Cl2N2,CAS号1910-42-5,纯度大于或等于99.0%。
3.3.2 混合方法工作溶液
3.3.2.2将1000ug/mL的方法储备溶液稀释成0.2ug/mL。
3.3.2.3将0.2ug/mL的混合方法溶液分别移取1mL、2mL、4mL、5mL至4个10mL的容量瓶中,浓度依次为0.02 ug/mL、0.04 ug/mL、0.08 ug/mL、0.10 ug/mL、。
3.3.2.4方法混合溶液在0-4冰箱可保存一个月。
3.3.3基质混合方法工作溶液
基质混合方法工作溶液是将上述0.02 ug/mL、0.04 ug/mL、0.08 ug/mL、0.1ug/mL、0.2ug/mL的混合表准溶液分别移取0.5mL至5个棕色进样瓶中,再分别移取0.5mL样品空白基质提取液加入其中。最终五个点的浓度为0.01 ug/mL、0.02 ug/mL、0.04 ug/mL、0.05 ug/mL、0.1ug/mL。基质混合方法工作溶液应现配现用。
4仪器
4.1液相色谱-质谱/质谱仪:配有电喷雾(ESI)源。
4.2固相萃取装置。
4.3分析天平:感量0.0001g和0.01g。
4.4均质器。
4.5氮吹仪
4.6离心机:4000r/min。
4.7高速离心机:转速不低于10000r/min 。
4.8pH计。
4.9涡旋混合器。
4.10具塞离心管:50ml。
4.11容量瓶:50ml。
4.12移液管:10ml。
4.13刻度离心管:15ml。
5试样的制备与保存
5.1试样的制备
5.1.1 粮谷、豆类、棉籽和干木耳
取代表性样品500g,取可食部分,经磨碎机充分磨碎,混匀,装入洁净容器,密封并标明标记。
5.1.2 水果、蔬菜类
去壳、去籽、去皮、去茎、去根、去冠(不可用水洗涤),取水可食部分约500g,将其可食部分切碎后,依次用食品捣碎机将样品加工成浆状。混匀,均分成两份做为试样,分装如洁净的盛样袋内,密闭,表明标记。
5.2试样的保存
粮谷、豆类、棉籽和干木耳试样可于0~4保存,水果、蔬菜类试样于-18冷冻保存,在抽样及制样的操作过程中,应防止样品受到污染或者发生残留物含量的变化。
6 测定步骤
6.1 提取
称取约5g(精确至0.01g)试样于50mL具塞离心管中,加入25mL甲醇-0.1mol/L盐酸溶液(3.1.11),均质提取1min,4000r/min离心5min,取上层提取液至50ml容量瓶中,残留物再用20ml甲醇-0.1mol/L盐酸溶液重复提取一次,合并提取液于同一容量瓶中,并用水定容至刻度。准确移取10ml提取液,用1mol/L氢氧化钠溶液(3.1.7)调节pH值至7.0±0.1,并10000r/min离心5min,待净化。
6.2净化
将上述待净化液全部转移至经过预活化的Oasis WCX固相萃取柱中,控制流速在1ml/min-2ml/min,弃去流出液。依次用1ml水、1ml甲醇淋洗净化柱,最后用2ml乙腈-水-甲酸溶液洗脱,控制流速在1ml/min-2ml/min,收集洗脱液于15ml刻度离心管中,洗脱液经45氮吹仪吹干后,用1.0ml乙腈-0.1%甲酸溶液振荡溶解残渣,过0.22um滤膜后,供液相色谱-质谱/质谱仪测定。
6.3测定
6.3.1 液相色谱-质谱/质谱条件
液相色谱-质谱/质谱条件如下:
a) 色谱柱:Hilic柱,100mm,粒度1.7um,或相当者。
b) 流动相:
A:乙腈,B:0.1%甲酸溶液,梯度洗脱程序见表1.
表1 梯度洗脱程序表
| 时间 min | 流动相A % | 流动相B % |
| 0.25 | 80 | 20 |
| 1.00 | 80 | 20 |
| 1.50 | 20 | 80 |
| 2.00 | 20 | 80 |
| 2.50 | 80 | 20 |
| 3.50 | 80 | 20 |
c) 流速:0.25ml/min。
d) 柱温:30。
e) 进样量:5μL。
f)质谱条件:参见附录A。
6.3.2 液相色谱-质谱/质谱检测
| 相对离子丰度/% | >50 | >20-50 | >10-20 | ≤10 |
| 允许的相对偏差/% | ±20 | ±25 | ±30 | ±50 |
X= ……………………………………(1)
| 序号 | 中文名 | 加标量/(mg/kg) | 平均回收率/% | 相对方法偏差RSD/% | ||||||
| 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 | ||
| 1 | 敌草快 | 0.02 | 0.10 | 0.15 | 105.00% | 87.00% | 86.00% | 10.80% | 5.45% | 5.17% |
| 2 | 百草枯 | 0.02 | 0.10 | 0.15 | 90.8% | 88.6% | 85.4% | 5.76% | 4.35% | 4.01% |
表3 敌草快、百草枯在大米基质中添加回收率
附录A
质谱条件和多反应监测条件
| 电离方式 | ESI+ |
| 毛细管电压 | 3.0kV |
| 源温度 | 110℃ |
| 干燥气温度 | 350℃ |
| 干燥气流速 | 50L/h |
| 鞘气温度 | 250℃ |
| 碰撞电压 | 100V |
| 监测模式 | 多反应监测 |
表A.2 多反应监测条件
| 化合物 | 母离子 | 子离子 | 驻留时间 s | 碰撞能量 eV |
| 敌草快 | 183.1 | 156.9* | 0.10 | 20 |
| 183.1 | 129.9 | 0.10 | 30 | |
| 百草枯 | 171 | 77.2* | 0.10 | 19 |
| 171 | 154.9 | 0.10 | 26 | |
| 注:加“*”的离子用于定量。 | ||||
附录B
方法物质色谱图
图B敌草快、百草枯液相色谱-质谱/质谱多反应监测色谱图
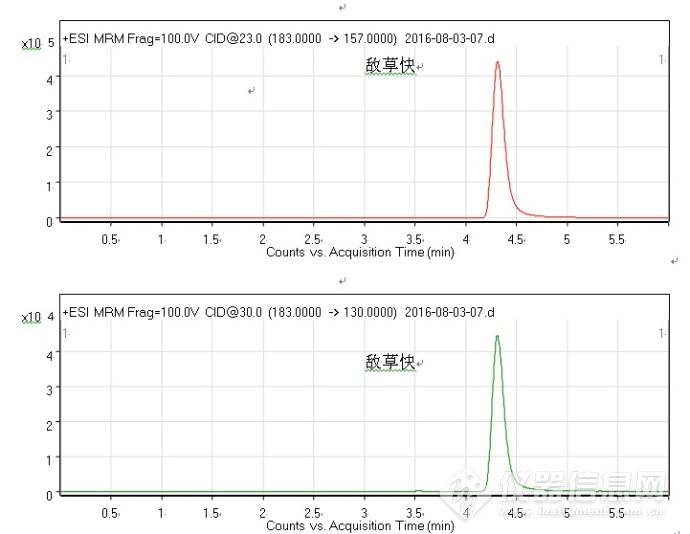 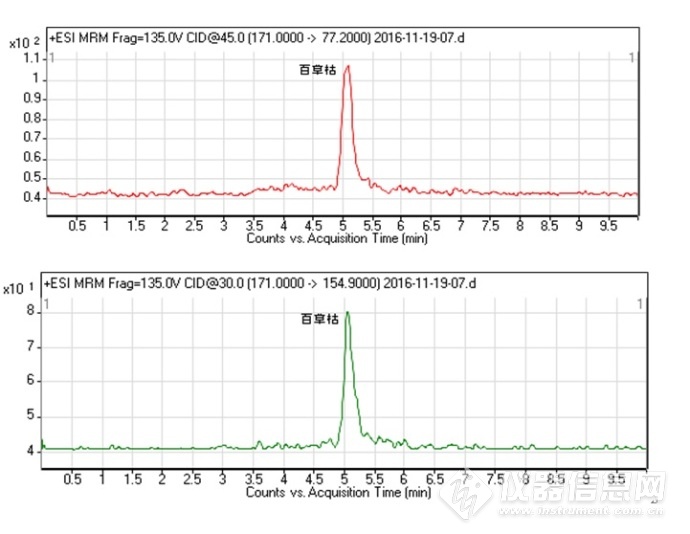 | |
附录C
敌草快、百草枯精密度数据表
| 序号 | 中文名 | 含量/ug/ml | 相对方法偏差RSD/% | |||||
| 1 | 敌草快 | 0.0197 | 0.0202 | 0.0202 | 0.0192 | 0.0201 | 0.0203 | 2.12% |
| 2 | 百草枯 | 0.0794 | 0.0782 | 0.0786 | 0.0781 | 0.0770 | 0.0768 | 1.29% |

































