推荐厂家
暂无
暂无

问题:暑湿感冒颗粒中5-O-甲基维斯阿米醇苷、升麻素苷的检测对照品分析中5-O-甲基维斯阿米醇苷的理论塔板数是?答案:24128.072【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币zengzhengce163(注册ID:zengzhengce163)ZHAOGUANGXI(注册ID:ZHAOGUANGXI)caishendao(注册ID:caishendao)http://ng1.17img.cn/bbsfiles/images/2016/03/201603021507_585734_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/03/201603021507_585735_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================暑湿感冒颗粒中5-O-甲基维斯阿米醇苷、升麻素苷的检测样品制备 制备方法1. 对照品:取5-O-甲基维斯阿米醇苷对照品、升麻素苷对照品适量,精密称定,加甲醇制成每1 mL各含20 μg的溶液,即得。2. 供试品:溶液取装量差异项下的本品,研细,取约4 g,精密称定,置具塞锥形瓶中,精密加入甲醇50 mL,称定重量,加热回流2小时,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。分析条件 色谱柱Diamonsil C18 150 x 4.6 mm,5 μm (Cat#:99901)流动相A:甲醇 B:水 梯度流速1 mL/min柱温30 ℃检测器UV 290 nm 进样量10 μL 色谱图对照品 http://ng1.17img.cn/bbsfiles/images/2016/03/201603020958_585675_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 26.320 311583 8810 12825.336 0.927 -- 2 49.733 341906 7082 24128.072 0.905 21.185 *药典要求理论板数按5-O-甲基维斯阿米醇苷峰计算应不低于5000供试品 http://ng1.17img.cn/bbsfiles/images/2016/03/201603020959_585676_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 26.289 39166 1313 16830.923 0.935 -- 2 49.737 62730 1428 25652.736 0.937 22.846 *药典要求理论板数按5-O-甲基维斯阿米醇苷峰计算应不低于5000
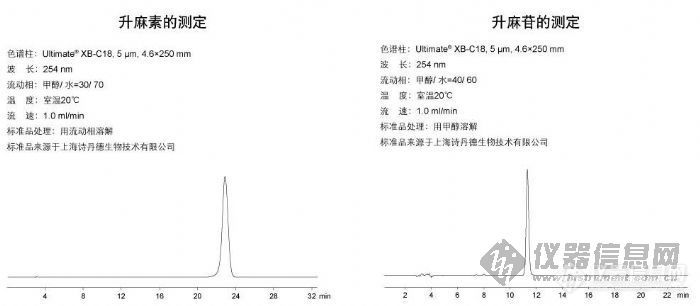
升麻素的测定和升麻苷的测定http://ng1.17img.cn/bbsfiles/images/2009/10/200910130312_175404_1896702_3.jpg
防风Saposhnikoviae Radix为伞形科植物防风Saposhnikovia divaricata (Turcz.) Schischk.的干燥根,具有祛风解表、胜湿止痛、止痉的功效[1]。防风含有多种化学成分,主要包括色原酮类、香豆素类、多糖类、挥发油类等[2],这些成分具有解热、镇痛、抗炎、抗菌、抗氧化、抗肿瘤等多种药理作用[3-6]。升麻素苷、升麻素、5-O-甲基维斯阿米醇苷为主的色原酮类成分是防风的主要活性物质,具有解热、镇痛、抗炎等多种药理活性[7-10],其中升麻素苷、5-O-甲基维斯阿米醇苷已作为《中国药典》2020年版中防风质量控制的标志物。课题组前期已对5-O-甲基维斯阿米醇苷体内外的代谢进行了全面的研究[11],但尚未见对升麻素苷的研究报道。有研究表明升麻素苷是防风抗炎作用的主要成分[12],而防风为《中国药典》2020年版收录的中成药齿痛消炎灵颗粒的君药,齿痛消炎灵颗粒具有治疗牙周炎的作用[13-14],故本研究采用超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]-四极杆-飞行时间质谱联用技术(UPLC-Q-TOF-MS/MS)与MetabolitePilot 2.0、PeakView 2.0软件,对升麻素苷在牙周炎模型大鼠和正常大鼠的体内外代谢进行研究,并比较代谢差异,为防风的药效物质基础提供依据。 1 材料 1.1 动物 SPF级雄性SD大鼠,体质量(220±20)g,由河北石家庄伊维沃生物技术有限公司提供,动物生产许可证号SYXK(冀)2020-002。动物于温度(22±2)℃、相对湿度(50±3)%、12 h光照/12 h黑暗循环环境下饲养。动物实验通过河北医科大学动物伦理委员会的伦理审查(批准号DW2019003)。 1.2 药品与试剂 升麻素苷对照品(批号BD121316,质量分数≥98%),购自上海毕得医药科技股份有限公司;升麻素对照品(批号HR1638W2)购自宝鸡市辰光生物科技有限公司;右美沙芬对照品(批号Y03S11W120802,质量分数>98%)购自上海源叶生物科技股份有限公司;甲醇、乙腈(色谱纯)购自美国Tedia公司;甲酸(色谱纯)购自美国Diamond公司;纯净水购自娃哈哈有限公司。 1.3 仪器 Triple TOF 5600+型高分辨质谱仪(美国AB Sciex公司);超高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]系统包括CBM20A型控制器、DGU-20A5R型在线脱气模块、LC-30AD型二元梯度高压泵系统、SIL-30AC型自动进样器和CTO-30A型柱温箱(日本岛津公司);D3024R型高速冷冻离心机(美国SCILOGEX公司);KQ-5200E型台式超声波清洗器(昆山市超声仪器有限公司);十万分之一分析天平(瑞士METTLER TOLEDO公司);MTN-2800D型氮吹仪(天津奥特赛恩斯仪器有限公司);MX-S型涡旋混匀器[大龙兴创实验仪器(北京)有限公司];SkyScan 1176型小动物Micro-CT扫描影像系统、NRecon软件、3D重建软件CTvox(德国Bruker公司)。 1.4 数据处理软件 Analyst® TF 1.7软件、MetabolitePilot 2.0软件、PeakView 2.0软件(美国AB Sciex公司)。 2 方法 2.1 溶液的制备 2.1.1对照品溶液的制备 精密称取升麻素苷、升麻素对照品适量,甲醇溶解制成质量浓度为1 mg/mL的贮备液,并用甲醇稀释成质量浓度为100 μg/mL的对照品溶液。 2.1.2内标(IS)溶液的配制 精密称取右美沙芬对照品适量,甲醇溶解制成质量浓度为1 mg/mL的贮备液,临用前用甲醇稀释成质量浓度为2.0 μg/mL的IS溶液。 2.1.3大鼠ig溶液的配制 精密称取升麻素苷对照品600 mg,加入60 mL纯净水,配成质量浓度为10 mg/mL的ig溶液。 2.2 牙周炎模型的建立 大鼠适应性喂养1周,采用吸入式2%~3%异氟烷实施麻醉,麻醉成功后采用0.2 mm正畸结扎丝结扎大鼠上颌左侧第一磨牙牙颈部,诱导实验性牙周炎[15-17],对侧同名牙作为对照不结扎。手术后,待大鼠全部苏醒,喂其常规鼠粮及水,并定期检查结扎丝牢固程度,整个实验持续8周。 在整个实验过程,注意大鼠精神状态,定期检查牙龈炎症、颜色、水肿、探针是否出血及牙周袋是否形成等。造摸8周后随机抽取牙周炎模型大鼠,处死,取上颌左侧第一磨牙及牙龈组织,同时取其对侧对比,置于4%多聚甲醛固定,常规脱钙,脱水透明,浸蜡包埋,制作切片,常规苏木素-伊红(HE)染色后,于显微镜下观察病理变化。同时采用微型计算机断层成像技术(micro computed tomography,Micro-CT)重建大鼠牙槽骨三维图像,观察牙槽骨吸收情况。 2.3 动物给药及生物样品采集 实验前,将正常大鼠随机分为4组,每组3只,第1组为空白正常胆汁组,第2组为给药正常胆汁组,第3组为空白正常血浆、尿液及粪便组,第4组为给药正常血浆、尿液及粪便组。模型组按与正常组相同的方法随机分为4组。给药前大鼠禁食12 h,自由饮水。根据大鼠体质量,以100 mg/kg的剂量进行ig给药。空白组ig等体积的纯净水溶液。 分别于ig给药后0.167、0.333、0.5、0.75、1、2、4、6、8、12、24 h自眼内眦取血0.3 mL,置肝素化的离心管中,4 ℃、3 500 r/min离心10 min,取上清液获得血浆样品,并将各组不同时间点获得的血浆样品合并,即得空白血浆和含药血浆。ig给药后,收集大鼠0~72 h每4个小时的尿液和粪便样本,并将来自同组大鼠的所有尿液和粪便分别进行混合,即得空白和给药尿液、粪便。ig给药后,通过ip 20%乌拉坦生理盐水溶液(1.5~2.0 g/kg)麻醉,实施胆汁插管引流手术收集0~24 h的胆汁样品,将来自同组大鼠的所有胆汁进行混合,即得空白和给药胆汁。所有样品置于?80 ℃冰箱备用。 分别取正常大鼠及牙周炎模型大鼠新鲜粪便3 g,加入30 mL厌氧培养液[18],用玻璃棒研碎并搅拌均匀,经医用纱布滤过,即得肠道菌培养液。取肠道菌培养液1 mL,加入100 μL(1.0 mg/mL)升麻素苷溶液,通入氮气置于无氧并充满氮气的厌氧袋中,在提前预热至37 ℃的摇床中孵育12 h,即得肠道菌孵育样品。空白组用100 μL超纯水代替升麻素苷溶液。 2.4 样品前处理 取血浆、尿液、胆汁样品各1 mL,分别加入100 μL的IS溶液(2 μg/mL),混合均匀,加入3倍量甲醇,涡旋5 min,4 ℃、15 000 r/min离心10 min,取上清液。取肠道菌孵育液1 mL、粪便样品0.5 g(加1 mL蒸馏水超声30 min制备成匀浆),分别加入100 μL的IS溶液(2 μg/mL),加入3倍量的醋酸乙酯,涡旋5 min,4 ℃、15 000 r/min离心10 min,收集上层萃取液,重复萃取3次,合并萃取液。将离心后所得的各生物样品上清液置另一清洁离心管中,N2流吹干,200 μL 50%甲醇复溶,涡旋5 min,15 000 r/min离心10 min,取上清液即得。 2.5 检测条件 2.5.1 色谱条件 COSMOCORE C18柱(150 mm×2.1 mm,2.6 μm);流动相为0.1%甲酸水溶液(A)- 甲醇(B),梯度洗脱:1~3 min,5%~20% B;3~25 min,20%~95% B;25~30 min,95% B。预平衡5 min,柱温40 ℃,体积流量0.3 mL/min,进样量5 μL。 2.5.2 质谱条件 电喷雾离子源(electro-spray ionization,ESI),正离子模式下进行全扫描,参数设置如下:离子源喷雾电压5 500 V;源温度550 ℃;气帘气压力241.325 kPa;雾化气(Gas1)压力379.225 kPa;加热气(Gas2)压力379.225 kPa;解簇电压70 V;碰撞能量40 eV;碰撞能量扩展15 eV。TOF-MS扫描的扫描范围为m/z 100~1 200,积累时间设置为250 ms。每个扫描周期选择8个响应最高的离子进行MS/MS扫描。产物离子扫描范围为m/z 50~1 200,积累时间为100 ms。 2.6 数据处理 正常大鼠和牙周炎模型大鼠ig升麻素苷后,各生物样本的总离子流图见图1。采用以下5个步骤来鉴定和分析升麻素苷在正常大鼠及牙周炎模型大鼠体内外的代谢物:①基于UHPLC-Q-TOF-MS/MS技术,在线进行全扫描数据采集,并利用多重质量亏损(MMDF)和动态背景扣除(DBS)设置获得准确的MS/MS质谱信息。②利用Peak View、Metabolite Pilot软件中的多种数据挖掘工具,自动过滤出升麻素苷的可能代谢物。③从准确的质谱数据、母体药物的裂解模式以及相关文献描述代谢物的推断过程。④ClogP值用作区分具有相同分子式和相似质谱数据的代谢物异构体的参数。ClogP值越大,在反相色谱系统中的洗脱时间就越长。⑤根据Peak View软件提供的代谢物的峰面积,用峰面积相对定量法比较代谢物在正常大鼠及牙周炎模型大鼠各生物样品中的含量差异。3 结果 3.1 CT成像 应用Micro-CT扫描、三维图像重建显示,与对照组(图2-B、D)相比,模型组(图2-A、C)牙槽骨吸收明显,同时有水平和垂直向吸收。对大鼠上颌第一、第二磨牙兴趣区域[19](本实验选取的兴趣区域(region of interest,ROI)为第一、第二磨牙近中牙槽嵴吸收情况)进行测量,对照组牙近中釉牙骨质界(cemento-enamel juction,CEJ)到牙槽嵴顶(alveolar bone crest,ABC)的平均距离为0.394 mm(图2-D),与对照组相比,模型组CEJ到ABC的距离平均增至0.813 mm。分析大鼠CEJ至ABC的垂直距离(图2-A、B),即分别取对照组与模型组样本牙齿颊侧的近中、中央及远中共3个位点的CEJ至ABC的距离,测量统计分析结果显示,与对照组相比,模型组CEJ-ABC距离明显增加(P<0.05,图2-E)。 图片 3.2 升麻素苷质谱裂解规律分析 升麻素苷(M0,C22H28O11)的保留时间为8.80 min,M0通过重排生成准分子离子峰[M+H]+m/z469.170 2。母离子m/z 469.170 2失去18(-H2O)、72(-C4H8O)分别形成特征碎片离子m/z 451.173 2、397.125 0。M0通过丢失162(-glu)、234(-glu-C4H8O)分别产生特征碎片离子m/z307.128 6、235.068 0,其中特征碎片离子m/z 235.068 0是特征碎片离子m/z307.128 6通过二氢吡喃环失去C4H8O发生RDA裂解反应产生。通过连续丢失O和C3H6O后,m/z 235.068 0产生碎片离子m/z 219.072 5、161.065 4。苷元离子m/z307.126 8有2种脱水方式(-H2O),分别产生m/z289.116 6的2种结构不同碎片离子,苷元离子在失去水的基础上连续失去2个CH3分别产生m/z274.092 3、259.068 8。通过连续丢失C3H6、CH2、C2H2、CO和O后,m/z 289.116 6产生了一系列碎片离子m/z247.068 1、233.052 3、221.051 7、219.072 5、205.056 3、193.056 0、189.061 1、177.060 6。升麻素苷可能的裂解途径见图3。 3.3 升麻素苷在大鼠体内外代谢物的分析鉴定 采用上述分析策略,共鉴定出30个升麻素苷的代谢物(其中I相代谢物25个、II相代谢物5个)。在健康大鼠中,共发现25个代谢产物(血浆中8个、尿液中17个、粪便中11个、胆汁中19个、体外肠道菌群3个)。在牙周炎模型大鼠中,共发现27个代谢产物(血浆中8个、尿液中18个、粪便中12个、胆汁中22个、体外肠道菌中2个)。升麻素苷原型及30种代谢物的详细信息见表1。 3.3.1 I相代谢物的鉴定 (1)水解(M1):M1的保留时间为10.14 min,准分子离子为m/z 307.118 0,推测其分子式为C16H18O6。M1的相对分子质量比M0少162(C6H10O5),此外,M1的特征碎片离子m/z 307.118 0、289.107 4、274.085 0、259.059 5、247.061 9、235.059 4、233.044 1、221.043 9、217.050 3、205.049 6、189.055 0、177.054 3、161.059 2均与M0相同,这表明M1在M0基础上发生了糖基化反应。且经过对照品比对,确认M1是M0脱糖产生的水解产物升麻素。 (2)水解-羟基化:M2~M5的保留时间分别为5.98、7.12、7.92、8.65 min,准分子离子峰分别为m/z323.113 4、323.111 2、323.112 8、323.113 0,均比M1多16,表明M2~M5可能为M1的单羟基化产物,推测其是分子式C16H18O7的同分异构体。M2的特征碎片离子m/z323.113 4、305.101 9、275.085 0、263.064 4均比M1的特征碎片离子m/z307.118 0、289.107 4、259.059 5、247.061 9多16,此外M2的特征碎片离子m/z 247.060 2、235.059 4、233.046 2、221.044 1、177.054 9均与M1的特征碎片离子保持一致,尤其特征碎片m/z263.064 4的存在,说明该羟基化反应发生在C-2′位。M3的特征碎片离子m/z 323.111 2、305.109 0、275.055 4均比M1的特征碎片离子m/z 307.118 0、289.107 4、274.085 0、259.0595多16,且其特征碎片离子m/z 235.031 5、233.035 6、205.072 2、193.050 1均与M1的特征碎片保持一致,说明该羟基化反应可能发生在C-5′位或者C-2′位。M4的特征碎片离子m/z259.060 0、247.060 0、235.060 2、233.044 6、221.044 8、205.048 5、189.054 3、177.054 9、161.060 1与M1具有相同的裂解途径,且根据其特征碎片m/z 305.1015的存在,推测羟基化的位置可能在C-3′位。M5的碎片离子m/z323.113 0、305.102 6与M1相比增加了16,其特征碎片离子m/z 259.061 1、247.060 1、235.060 3、233.043 9、221.044 7、205.050 2、189.054 9、177.054 5均与M1具有相同的裂解途径,尤其特征碎片离子275.054 8的存在,推测M5的羟基化反应可能发生在C-8位上。通过计算ClogP值发现,M2~M5的ClogP值分别为?0.859 1、?0.756 5、?0.364 7、?0.018 9,与保留时间的大小具有一致性,证明了推测的合理性。 (3)水解后失去CH2:M6保留时间为12.85 min,准分子离子峰为m/z293.102 3,推测其分子式为C15H16O6,是在M1的基础上丢失1个CH2。M6主要的二级碎片离子m/z 293.102 3、275.091 6、245.043 7、221.044 8均比M1的碎片离子m/z307.118 0、289.107 4、274.085 0、259.059 5、235.059 4少14,且由于特征碎片m/z 275.091 6、233.044 7的存在,提示反应位点可能在C-5。 (4)水解后失去CH2+O:M7、M8的保留时间分别为7.19、9.67 min,准分子离子峰分别为m/z 309.096 6、309.097 7,推测其分子式为C15H16O7。与M6相比多了16。因此,代谢物M7、M8被初步鉴定为M6的羟基化产物。M7的特征碎片离子m/z233.026 8、221.029 1、205.031 9均与M6保持一致,且其特征碎片离子m/z309.096 6、291.099 1与M6相比增加了16,提示羟基化的位置发生在侧链上,推测反应位点在C-5′位。M8的特征碎片m/z 233.050 8、221.045 6、177.083 7均与M6的特征碎片离子保持一致,说明M8与M6具有相同的裂解途径,且有特征碎片m/z 249.164 1的存在,说明羟基化的位置在环上,推测反应位点可能在C-8位。此外,根据上述所推结构,计算M7、M8的ClogP值分别为?0.320 8、0.337 1,与出峰时间保持一致,证明了推测的合理性。 (5)水解后去甲基化成羧酸:M9、M10的保留时间分别为7.78、9.30 min,准分子离子峰分别为m/z337.092 9、337.091 9,推测其分子式为C16H16O8。与M1相比多了30,推测M9、M10是M1结构中的1个甲基被氧化成羧酸后得到的代谢产物。根据M1的结构特征推测发生反应的位点可能是与C-5连接的甲氧基及C-5′位的甲基上。M9的特征碎片离子m/z 337.092 9、319.082 3、304.060 0、289.032 9、277.071 1、265.032 2、263.056 3、235.024 3、219.029 1、207.043 8均比M1的特征碎片离子离子m/z 307.118 0、289.107 4、274.085 0、259.059 5、247.061 9、235.059 4、233.044 1、205.049 6、189.055 0、177.054 3多30,因此证明M9氧化位点发生在C-5位连接的甲氧基上。M10在正离子模式下形成的准分子离子峰为m/z 337.091 9,与M1相比多30,其特征碎片离子m/z274.130 1、259.063 0、247.060 7、235.061 7、233.045 8、205.051 7、161.061 8均与M1的特征碎片离子保持一致,说明其氧化位点发生在M1的侧链上而不在环上,故推测反应位点发生在C-5′位。根据以上推测的结构,计算两者的ClogP值分别为?1.233 0、?0.585 6。计算结果与出峰时间顺序保持一致,进一步确证了推测的合理性。 (6)水解后去甲基化成酮:M11保留时间为10.96 min,准分子离子峰为m/z 291.086 3,推测其分子式为C15H14O6。与M1相比少了16,推测在M1的基础上失去了1个甲基进一步将连接在中心碳原子上的羟基氧化成酮。M11的特征碎片离子m/z 291.086 3、273.076 7、258.058 0与M1相比均少16,特征碎片离子m/z 259.062 8、235.096 5、233.044 7、221.041 4、177.052 1与M1保持一致,说明反应发生在侧链上,因此推测反应发生在C-4′位。 (7)水解脱羟基:M12的保留时间为9.83 min,其准分子离子峰m/z 291.122 6,推测其分子式为C16H18O5。与M1相比少了16(O),推测M12在M1的基础上发生了脱羟基反应,M12的特征碎片离子m/z 273.076 0、263.092 2、219.025 6、217.159 5均比M1的特征碎片离子m/z 289.107 4、247.061 9、235.059 4、233.044 1少16,说明脱羟基位发生在C-9位。 (8)水解后失去CH2O:M13的保留时间为8.24 min,其准分子离子峰为m/z277.106 7,推测其分子式为C15H16O5。与M1相比少了30(CH2O),推测M13可能是在M1的基础上丢失甲氧基产生的代谢物。M13的特征碎片离子m/z 259.101 8、217.054 2、205.052 8、181.089 7可能是M1的特征碎片离子m/z289.107 4、247.061 9、235.059 4、221.043 9丢失1个甲氧基产生的,根据M1的结构特点推测丢失位点在C-5位。 (9)水解后脱羟基失去CH2O:M14的保留时间6.48 min,其准分子离子峰为m/z261.112 5,推测其分子式为C15H16O4。与M12相比少了16,与M1相比少了46(M1-O-CH2O),推测M14可能是在M1的基础上先失去羟基又失去甲氧基产生的。M14的特征碎片离子m/z 243.215 1、228.206 8、189.090 3与M1的特征碎片离子m/z 289.107 4、274.085 0、235.059 4相比均少46,且M14的特征碎片离子m/z243.215 1正好比M12的特征碎片离子少16,证明推测合理。 (10)水解脱羟基后被氧化成醛:M15的保留时间为8.23 min,其准分子离子峰为m/z 305.102 4,推测其分子式为C16H16O6。M15的特征碎片离子m/z 287.132 8比M12的特征碎片离子m/z 273.076 0多14,水解后脱羟基位点与M12保持一致。且其特征碎片离子m/z290.071 9、275.054 4、247.058 3、233.044 4可能是在M15的准分子离子m/z 305.102 4的基础上通过连续丢失2个甲基、CO和亚甲基产生的。因此,推测M15是代谢物M12中的1个甲基被氧化成醛产生的。根据母药结构特点,结合π-π共轭体系可能使结构更加稳定的规律,推测氧化反应位点最可能发生在2位的CH3上。 (11)失去C4H8O:M16的保留时间为8.80 min,其准分子离子峰为m/z397.113 6,推测其分子式为C18H20O10。与M0相比少了72,根据其结构特点推测M16可能是M0失去C4H8O所得到的代谢产物,其特征碎片m/z 235.05 51、205.136 9与M0保持一致,且特征碎片m/z72.086 6(-C4H8O)的存在,初步认为推测M16的结构合理。 (12)水解脱羟基后去甲基成羧酸:M17的保留时间为11.08 min,其准分子离子峰为m/z 321.097 4,推测其分子式为C16H16O7。与M9相比少了16,与M1相比多了14,推测M17可能是M1脱羟基后的1个甲基被氧化成羧酸的产物。其特征碎片离子m/z 321.097 4、303.085 9正好比M1的特征碎片离子m/z289.107 4多14,比M9的特征碎片离子m/z 319.082 3少16,特征碎片离子m/z273.039 5比M9的特征碎片离子m/z 289.032 9少16,且其含有特征碎片离子m/z 235.022 7、205.049 6,与M1和M9一致。说明M17与M9结构相似,氧化位点与M9一致。 (13)单羟基化反应:M18~M20的保留时间分别为6.24、6.93、9.00 min,其准分子离子峰分别为m/z 485.166 2、485.166 5、485.164 7,推测其分子式为C22H28O12。与M0相比多了16(O),因此推测他们可能是M0的单羟基化产物。根据母药的结构特点可以看出羟基化反应发生在C-3′、C-5′和C-8位时相对稳定。M18的特征碎片离子m/z247.060 0、235.058 3、233.043 8、205.048 4均与M1的特征碎片离子保持一致,且其特征碎片






 留言咨询
留言咨询
 留言咨询
留言咨询
 留言咨询
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP


