

方案详情文
智能文字提取功能测试中
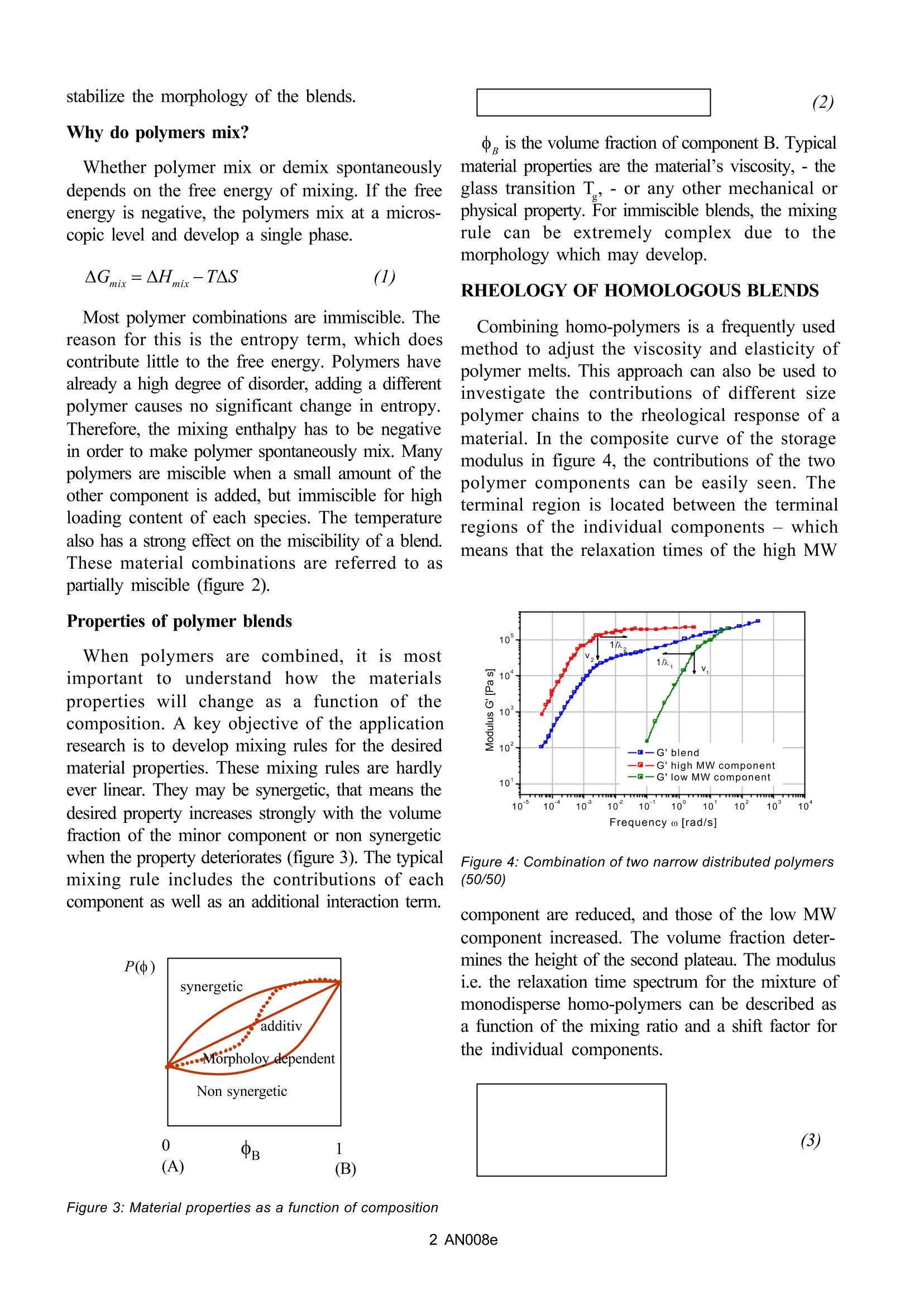
AN008e Mixing Rules for Complex Polymer Systems A. Franck, TA Instruments Germany Keywords:Mixing rules, polymer blends, miscibility, multi-phase systems INTRODUCTION TO THE BEHAVIOR OFPOLYMER BLEND SYSTEMS Complex polymer systems are combinations ofdifferent types of polymers which can exist as singleor multi-phase systems. The polymers of thecomponents are chosen according to criteria like:-costs,-processing performance,-mechanicalproperties, - thermal properties, etc.. One of the mainreasonfsor combining polymers is effectively costs.Polymer combinations are cost effective sincemixing an expensive material with a less expensiveone, provides increased performance at a lower price.Polymers are also blended to combine the specificproperties of different materials in one. Crystallineand amorphous materials are good combinations.Amorphous materials are transparent and have abetter dimensional stability; crystalline materials arestiffer. The combination of a thermoplastic materialwith an elastomer provides high stiffness and good e( Figure 1: Combinations of polymer materials. impact resistance. Combi-ning polymer materialspermits to create new material properties, withoutdeveloping a new material. HOW TO DEVELOP COMPLEX POLYMERCOMBINATIONS? A simple approach to modify polymers is toincorporate solid particles or fibers. Thesecomponents act as reinforcements and increase thestrength of the material Combining chemical different types of polymersin a melt mixing process to form a polymer blendhowever increases considerably the possible ma-terial and property variations. Material combinationscan be miscible or immiscible (Figure 1). Themiscible blends can be combinations of polymersof the same kind (homologous) or of different nature(heterogeneous). Immiscible polymer combinationsare multiphase systems. The morphology plays animportant role and significantly influences the finalmaterial properties. Properties can be modified usingcompatibilizers such as co-polymers to change and Figure 2:Gibbs free energy for miscible and immisciblepolymer combinations Why do polymers mix? Whether polymer mix or demix spontaneouslydepends on the free energy of mixing. If the freeenergy is negative, the polymers mix at a micros-copic level and develop a single phase. Most polymer combinations are immiscible. Thereason for this is the entropy term, which doescontribute little to the free energy. Polymers havealready a high degree of disorder, adding a differentpolymer causes no significant change in entropy.ThTherefore, the mixing enthalpy has to be negativein order to make polymer spontaneously mix. Manypolymers are miscible when a small amount of theother component is added, but immiscible for highloading content of each species. The temperaturealso has a strong effect on the miscibility of a blend.These material combinations are referred to aspartially miscible (figure 2). Properties of polymer blends When polymers are combined, itismostimportant to understand how the rmatproperties will change as a funnccttiioonn of tcomposition. A key objective of the applicationresearch is to develop mixing rules for the desiredmaterial properties. These mixing rules are hardlyever linear. They may be synergetic, that means thedesired property increases strongly with the volumefraction of the minor component or non synergeticwhen the property deteriorates (figure 3). The typicalmixing rule includes the contributions of eachcomponent as well as an additional interaction term. Po) e , is the volume fraction of component B. Typicalmaterial properties are the material’s viscosity,-theglass transition T-or any other mechanical orphysical property. For immiscible blends, the mixingrule can be extremely complex due to themorphology which may develop. RHEOLOGY OF HOMOLOGOUS BLENDS Combining homo-polymers is a frequently usedmethod to adjust the viscosity and elasticity ofpolymer melts. This approach can also be used toinvestigate the contributions of different sizepolymer chains to the rheological response of amaterial. In the composite curve of the storagemodulus in figure 4, the contributions of the twopolymer components can be easily seen. Theterminal region is located between the terminalregions of the individual components - whichmeans that the relaxation times of the high MW Figure 4: Combination of two narrow distributed polymers(50/50) component are reduced, and those of the low MWcomponent increased. The volume fraction deter-mines the height of the second plateau. The modulusi.e. the relaxation time spectrum for the mixture ofmonodisperse homo-polymers can be described asa function of the mixing ratio and a shift factor forthe individual components. For a binary blend with 中,, the volume fractionof the high molecular weight component, theformula for a simple linear and a quadratic mixing rule are given in (4) and (5). In order to check, thevalidity ofthese simple mixing rules, the weightfactor V, is plotted as a function of the weightfraction of the high molecular weight component.V,, has been obtained from the relaxation timespectrum, reduced to a simple box-distribution, asshown in the figure 5. /1/ It can be seen, that the Figure 6: Zero shear viscosity and equilibriumcompliance experimental data fall in-between the limits of thelinear and quadratic mixing rule- thus none of theserules describes the experimental findings. The shiftfactors aund A were found to be also dependent1on the weight fraction. In addition the higher MWshift factor A2 scales with M, which means thatthe high MW component has a stronger influenceon the rheology of the blend /1/. A good material property to check the mixing rule,is the zero shear viscosity n, and the equi-libriumcompliance J. Whereas the viscosity increases withthe weight fraction of the high molecular component中,, the equilibrium compli-ance goes through amaximum at low content of the high molecularweight component (Figure 6). Neither the linear orquadratic mixingrules describe the equilibriumcompliance. Based on the reptation theory/2,3/ Haley hasderived a mixing rule for the modulus and theviscosity as follows: The reptation model is quadratic in the weightfraction and includes a complex interaction term. tand t, are the monodisperse relaxation (reptation)times for the two components of the blend /2/. isthe local friction factor and M the entanglementmolecular weight. The viscosity and the equilibrium compliance asa function of the high molecular weight componentare much better predicted by this modified mixingrule and shown in figure 7. The mixing rule from Tsenoglou has beenextended /4/ and the weight fraction of the highmolecular component replaced by the molecularweight distribution w(M). The extended mixing law is used to obtain the MWD from rheological data.Figure 8 shows the MWD for a bimodal distributionblend of two PS with different molecular weight,extracted from the relaxation time spectrum i.e.dynamic data G’and G/5/. For reference, SECdata have been plotted also. Good agreementbetween the results from SEC and Rheology havebeen obtained. Volume fraction o。 Figure 6: Zero shear and equilibrium compliance RHEOLOGY OF HETEROGENEOUSMISCIBLE POLYMER BLENDS Heterogeneous polymer systems are composedof polymers of different chemical nature and glasstransitions T. Completely miscible heterogeneousblends exhibit only one T, which is located betweenthe Tof the pure components. Typical miscibleblends are PS/PVME (Polystyrene/Poly(vinylmethyl ether)), PSAN/PMMA (Poly(styrene s-acrylnitrile)/Poly(methyl methacrylate)), PEO/PMMA (Poly(ethylene oxide)/Poly(methyl meth-acrylate)), PB/PIP (Polybutadiene/Polyiso-prene),etc.. Figure 8: MWD determined from Rheology and SEC Miscible heterogeneous blends have been foundnot to scale with the mixing rules developed forhomologous polymer combinations. Also miscibleblends have only one Tg l i k e h o m o p o l y m e r s , t h e Tgis a strong function of the composition, oftenasymmetric and broader than the T of the purecompnents. Heterogeneous blends furthermore donot follow the time temperature superposition andas such are thermo-rheological complex. Due to the connectivity of the monomers alongthe main chain, local self concentration of thechemical different components leads to con-centration fluctuations in the fluid phase /6/ as shownin figure 9. The self-concentration is defined as thevolume of a Kuhn segment divided by the volumegiven by the length’of one Kuhn segment. Sincepolymers with a low T are more flexible, they havea higher self concentration ( for PI=0.45). As aresult of the self concentration effect, the polymercomponents locally behave more like in the purepolymer and the segmental dynamics in the blendtherefore are not only temperature, but alsodependent on concentration. Figure 10: Experimental and effective glass transition asa function of the volume fraction The figure 10 shows the experimental T() forthe PMMA/PSAN blend obtained from DSCmeasurements. The envelop curves represent theeffective glass transitions Teff which are the T atthe effective local composition bef adjusted for theself concentration中. Since low T components are more flexible, theself concentration , is higher and the localdynamics in the mixture are more like in the purecomponent;-the local Teff being suppressed incomparison to the experimental T ogf the mixture. HighT components have a lower and the localdynamics in the mixture are more representative ofthe dynamics of the mixture itself. Therefore the Teffollows much closer the T of the mixture. Due tothe local variation of T with the volume fraction,the experimental T broadens as the concentrationof PSAN decreases in the mixture. As a consequence of the local concentrationfluctuations, the local friction coefficient varies withthe volume fraction also. is now a function not only of temperature, butalso of the relative concentration of the components.The representative relaxation (reptation) time(equation 8) /7/ for each component has to be corrected for the friction factor. The M molecularweight at the volume fractiono is calculated fromthe individual components as follows: The time constants for the component A and Bin the mixing rule, i.e. the mixing rule fromTsenoglou, developed for the homologous blends,can now be replaced and the rheological behaviorof the blend as a function of the relative com-position calculated. The viscosity for the PMMA/PEO blend (figure 11) has been predicted for a selfconcentration of 中 =0.6. PEO is a small monomerand as such quite flexible, which is reflected in thehigh self concentration value /8/. RHEOLOGYOF IMMISCIBLE BLENDS Most polymersystems are not compatible and assuch immiscible. Their blends are multiphasesystems and as such often have a complexmorphology. In order to obtain for example thedesired mechanical properties, these blends need tobe modified further using a co-polymer to improvethe adhesion at the interface. Both, compatibalizersand the flow during processing are important tochange and stabilize the morphology -thusimproving the physical properties of these complexpolymer systems. The> properties control-led byblending include surface properties, impactresistance, thermal properties, dimensional stability,gas barrier, and ease of processing. Figure 11: Experimental and calculated viscosity as afunction of the components Dynamic mechanical response of a PS/PMMAblend A good example of an immiscible blend is the PS/PMMA combination. In the following study /9/PMMA is the continuous phase and PS is thedispersed phase in form of spherical inclusions. Theblend was prepared in a melt mixing process usingan extruder. The PS and PMMA samples were chosen to havesimilar rheological properties in the frequencyresponse (similar relaxation time spectrum) To characterize the blends, frequency dependentoscillation measurements on samples of 5, 10 and20% of PS as the dispersed phase have beenp1s G‘ierformed. The storage modulus G in figure 12shows an additional relaxation at low frequency,which increases with the concentration of thedispersed phase.The magnitude of the additional lowfrequency relaxation seems to correlate with thevolume fraction of the minor phase. EM (Transmission Electron Microscopy)pictures show an increase of the size of the spheresof the dispersed phase as well as a broadening ofthe distribution of particle sizes with increasing PSconcentration. The extensive increase of theelasticity of the blend, due to a higher G’contri-bution at low frequency in comparison to the pure components, has to be attributed to energy storagemechanisms of the morphology itself. Mechanicalenergy is stored as interfacial energy while thespherical inclusionsare deformed, and then dissipated as they recover to the original sphericalshape. Since the PS domain relaxation is slow, it canbe separated from of the fast relaxation of thecomponents. The energy storage mechanism is theinterface tension; the energy dissipation mechanismis the friction at the interface. How can these experimental findings be modeledand accounted for in mixing rules to predict therheological behavior of immiscible blends? Considerthe simplest multiphase system, which is a hardsphere in a Newtonian fluid. According to Einstein/10/ the viscosity increase in such a system is ahydrodynamic effect and depends on the volumefraction of the added particles only. If the solidparticles are replaced by deformable droplets of aNewtonian fluid, the system exhibits a viscoelasticresponse with a relaxation time which depends onthe viscosity of the continuous phase and theinterface tension. A simple model for the viscosityand the normal stress for an emulsion of twoNewtonian fluids has been derived by Choi andSchowalter /11/. For immiscible polymer systems,the mixing rule can be modified to include the phaserelaxation phenomenon as: For the PMMA/PS blends investigated, therelaxation of the phase is much slower, then the component relaxation.Gramespacher et al. /12/ extracted the re-laxation spectrum fromthe experimental mo-duli G’and G” andplotted the weightedspectrum tH(lnt) vs.the relaxation time asshown in figure 13. Thecharacteristicphaserelaxation time wasdetermined from thesecond maximum ofthe spectrum and the inter-face tension I was deter-mined from: Here a represents the drop size. Note: in order toobtain the average particle size, the interface tensionmust be known and the interfacial tension can be calculated if the average particle size is known. Monitoring the change of particle size duringflow using rheology Immiscible blend are difficult to mix at amicroscopic level. Mechanical energy has to beintroduced to mix the components and to offsetcoalescence. The resultant droplet size is a functionof the energy input, the applied flow conditions, etc..Can rheology be used to monitor the evolution ofparticle size under defined flow conditions? Vinckier et al./13/ have studied the immiscible Figure 13: Relaxation spectrum of the PMMA/PS blendand the pure componente blend of PDMS and PIB (70/30) in oscillation as afunction of pre-shear conditions. In figure 14,thedynamic modulus G’ at low frequency has amaximum at the lowest shear rates for the additionalphase relaxation. Under these conditions thedispersed PIB droplets are large and the interfacecan store a lot of mechanical energy. With increasingpre-shear rate the elastic contributions decrease; thishas to be interpreted as a break-up of the particles.Palierne /14 / proposed a emulsion model which isan extension of the Choi & Schowalter model toviscoelastic fluids. Graebling /15 / derived followingexpression for the average particle size: Figure 15 shows the excess relaxation spectrumextracted from the oscillation data in figure 14. Inorder to emphasize the effect of the phaserelaxation, the contributions for the componentrelaxations have been subtracted, assuming a linear mixing rule. The average droplet size shown in figure15 is calculated from the droplet relaxation time(peak values) using equation 14. The drop size Figure 14: Oscillation data for a 70/30 PDMS/PIB blendobtained decreases by one order of magnitude dueto droplet break-up. In order to follow the droplet break-up in real time,Vinckier et al. /16/ measured the transient viscosityand normal stress in start up and step up experiments(figure 16). In a start up or step up experiment, theviscosity first goes through a maximum, thendecreases to a minimum to reach a higher steadystate value again. Similar the normal stress showsan overshoot followed by a steady state. Startingfrom a spherical shape, the dispersed droplets areelongated, the viscosity decreases (less flowresistance), but the elasticity increases due to energy A-1va Figure 15: Excess relaxation spectrum and calculateddrop size for the PDMS/PIB blend as a function of pre- Fiugure 16: Transient viscosity and normal stress of aPDMS/PIB blend during start-up storage in the interface. At the maximum of thenormal stress, the droplets start to break up, theelasticity now decreases, however the viscosity, dueto the increase of small spherical droplets increasesagain.The steady state is a dynamic equilibrium statewith an average droplet size. Since the rheology depends on the morphologyand the morphology itself depends on the appliedflow conditions, the mixing law for the viscosity andelasticity of immiscible blends is very complex. Forthe PDMS/PIB blend, the zero shear viscosity isshown as a function of the concentration of PIB(figure 17). Only the conditions at low loadings ofeither PDMS or PIB can be described approximatelyby the Choi & Schowalter model/11/. The behaviorin the region of high volume fraction of bothcomponents is dominated by the morphology and Figure 17: Zero shear viscosity of a PDMS/PIB blends asa function of compositionj none of the existing models apply. Figure18 summarizes the results obtained on thePDMS/PIB blend. The normal stress Nand two times the storage modulus G’areplotted vs. shear rate or frequency. At lowfrequency i.e. low shear rate, oscillationand transient data for the elasticity (2Gand N,) of the blend superpose. Note, thatthe values for the pure components aresignificantly lower then for the blend. Atlow rate, the morphology in the transientexperiment is only slightly disturbed by Shearrate y [1/s]; frequency o [rad/s] Figure 18: Comparison of transient and oscillatoryexperiments conduted on a PDMS/PIB blend the flow and the large size droplets remains spherical.With increasing shear rate, the droplets are deformedand break down eventually. The normal stressremains high with increasing deformation asmechanical energy is stored in the interface. Inoscillation measurements, the long relaxation timesassociated with the phase relaxation do not respondto the high frequency probing. G’of the blenddecreases with frequency and reaches a valueslightly lower then the value of the pure componentof the continuous phase. This result is due to adilution effect of a small amount of the dispersedphase. CONCLUSION Mixing rules are important to predict the behaviorof multi-component polymer systems. The modifiedmixing rule from Tsenglou and Des Cloizeaux hasbecome an important analytical tool to determinemolecular weight distributions of homologouspolymers. The rheological properties of miscibleheterogeneous polymer systems are much more difficult to predict, nevertheless recent develop-ments provided a better understanding of thesesystems. In immiscible polymer systems, themorphology often dominates the rheologicalbehavior. The rheology of these systems is moredifficult to predict, especially as the morphology isalso strongly influenced by the flow history itself. REFERENCES ( /1/W.M. Prest Jr. Polymer J. 4,2(1973), 163 ) ( /2/ C. Tsenoglou , New Trends in Physics andPhysical Chemistry ofPolymers, 375 (1989) ) ( /3/J.C. Haley, et al. Macromolecules 36, 6142(2003) ) ( /4/ J.Des Cloizeaux, Macromolecules 23, 3992 (1990) ) ( /5/W.H. Tuminello p resented SOR, Oct 1999 ) ( /6/T.P. Lodge, T.C.McL e ish, Macromolecules 33, 5378(2000) ) ( /7/ R. J. Composto, E. J . Kramer, and D. M .White, Macromolecules 25,4167(1992) ) ( /8/ S. Wu, J.Polym.Sci.Part B Polym.Phys. 25, 2511(1987) ) ( /9/ C. Friedrich, e t alJ.Rheol. 93, 1411 ( 1995) ) ( /10/A. Einstein, A. Ann Phys. 19, 2 89 (1906),24, 591(1911) ) ( /1 1 / S.J . Choi , W.R. Schowalter Phys.Fluids 18,420(1975)(1975) ) ( /12/ Gramespacher et a l . J .Rheol. 36(6) 1 1 27(1992) ) ( /13/Vinckier et al . J.Rheol. 42(3), 705 (1997) ) ( /14/J.F. Palierne,Rheol. Acta 29, 204(1990) ) ( /15/D.Graebling et al. European PolymerJournal,30(3),301 (1994) ) ( /16/Vinckier et al. J.Rheol. 40(4), 613 ( 1996) ) AN Complex polymer systems are combinations of different types of polymers which can exist as single or multi-phase systems. The polymers of the components are chosen according to criteria like: - costs, - processing performance, - mechanical properties, - thermal properties, etc.. One of the main reasons for combining polymers is effectively costs. Polymer combinations are cost effective since mixing an expensive material with a less expensive one, provides increased performance at a lower price. Polymers are also blended to combine the specific properties of different materials in one. Crystalline and amorphous materials are good combinations. Amorphous materials are transparent and have a better dimensional stability; crystalline materials are stiffer. The combination of a thermoplastic material with an elastomer provides high stiffness and good impact resistance. Combining polymer materials permits to create new material properties, without developing a new material.
关闭-
1/9

-
2/9
还剩7页未读,是否继续阅读?
继续免费阅读全文产品配置单

TA仪器为您提供《复杂多组分聚合物中聚合物混合规则检测方案(流变仪)》,该方案主要用于聚合物基复合材料中聚合物混合规则检测,参考标准《暂无》,《复杂多组分聚合物中聚合物混合规则检测方案(流变仪)》用到的仪器有TA仪器ARES-G2 高级流变仪。
我要纠错
相关方案
 咨询
咨询







