

§张任-分析
第1楼2009/09/03
2\元素形态介绍
元素形态是指某一元素以不同的同位素组成、不同的电子组态或价态以及不同的分子结构等存在的特定形式。元素形态又分为物理形态和化学形态,其中物理形态是指元素在样品中的物理状态,如溶解态、胶体和颗粒状等;化学形态是指元素以某种离子或分子的形式存在,其中包括元素的价态、结合态、聚合态及其结构等。一般意义上所说的元素形态泛指化学形态,元素形态不同于元素价态,同一元素的相同价态可能有多种形态,如价态为五的砷元素,其元素形态可分为无机态和多种有机态的砷形态。不同元素的主要常见形态如表1所示:
表1不同元素的主要常见形态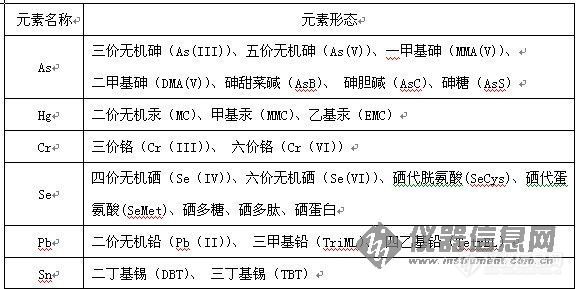
甲基汞的毒性要远高于无机汞,并且具有极强的生物亲和力,同时无机汞易于在生物体内富集并转化为甲基汞。人们首次认识到甲基汞的危害是在1955年,在日本的Minamata,因孕妇食用遭受甲基汞污染的鱼类,造成22名新生儿严重的脑损伤。在1971-1972年,伊拉克发生了大面积的甲基汞中毒事件,其原因在于当地人食用了经过甲基汞处理过的小麦种子做成的面粉。
Cr(III)是维持生物体内葡萄糖平衡以及脂肪蛋白质代谢的必需元素之一,而Cr(VI)却对生物体具有很大的毒性和致癌作用,原因在于其更强的氧化性和化学活性及迁移性;砷是一种有毒元素,但是不同形态砷的毒性却差别比较大,一般无机态砷毒性比较大,三价无机砷的毒性要大于五价无机砷;而有机态的砷中,甲基砷的毒性要强于其他的有机态砷,砷甜菜碱、砷胆碱和砷糖等则基本上没有毒性;对汞、锡和铅等重金属元素来说,有机态的化合物的毒性要远远高于无机态。作为人体必需的元素,铁仅仅是在二价时才能被生物体吸收和利用,食品中的总铁并不能代表可吸收利用的有效铁;硒是人体必需的元素,但是吸收过量时会导致硒中毒,不同形态硒的生物可利用性和毒性也差别较大;铝的毒性也和其形态密切相关,自由态的铝离子、水化羟基化合物Al(OH)2+和Al(OH)2+等是致毒形态,多核羟基铝也具有一定的毒性,而铝的氟配合物以及有机态配合物则基本无毒。
元素的不同存在形态决定了其在环境和生命过程中表现出不同的行为,不同的元素形态由于具有不同的物理化学性质和生物活性,在环境和生命科学领域发挥着不同的作用。元素总量或者浓度的相关信息已经不能满足环境、生命科学、毒理学等诸多领域研究的需要,有时候甚至会给出一些错误的信息。
§张任-分析
第2楼2009/09/03
3\元素形态分析及技术特点
根据传统分析方法所提供的元素总量的信息已经不能对某一元素的毒性、生物效应以及对环境的影响做出科学的评价,为此,分析工作者必须提供元素的不同存在形态的相关信息。元素形态具有多样性、易变性、迁移性等不同于常规分析对象的特点,因此其分析方法也成为一个崭新的研究领域,即“元素形态分析”。元素形态分析是分析科学领域中一个极其重要的研究方向,IUPAC将其定义为定量测定样品中一个或多个化学形态的过程。Lobinski将其定义为确定某一元素在样品中不同化学形态分布的过程;Caroli指出,形态分析是指识别和定量检测对人体健康和环境有危害的不同形态的无机分析物;Hieftje则将获得相关目标分析物原子的氧化态、键合特征、电荷态及原子缔合体的过程定义为形态分析;Welz则认为所谓元素形态分析是指测定特定条件下不同化合物的氧化态或可溶态的过程。曾有人根据Tessier连续萃取法将土壤中元素形态分为可交换态、碳酸盐结合态、铁-锰氧化物结合态、有机物结合态和残渣态等五种,但这并不是严格意义上的形态分析,这一萃取过程并不能提供涉及分子结构和电荷状态的元素形态的详细信息,仅能称之为“组分分析”。
Kolb在1966年,首次提出原子吸收光谱检测技术在作为气相色谱的检测器方面的潜在优势,开创了联用技术的先河。在20世纪70年代末至80年代初,Van Loon和Suzuki分别在权威期刊Anal. Chem.和Anal. Biochem.上发表了元素形态分析领域的开创性的工作,使得广大的分析工作者将其研究重点转移至元素形态分析技术的开发上来。经过近三十年的发展,元素形态分析已经成为分析科学领域的一个重要分支。随着这一技术的不断发展,已经为环境科学、生命科学、临床医学、营养学、毒理学、农业科学等领域提供了越来越多的有用信息。
由于一种元素存在几种甚至是几十种元素形态,且不同的元素形态间在一定的条件下会相互转化,因此元素形态的分析方法已不同于传统的总量分析。在前处理方法上需要保持元素的现有形态,因此也不能沿用传统的用于总量分析的酸消解方法和灰化法;在测定方法上,形态分析也远不同于传统的总量分析,对方法的检出能力和重复性提出了更高的要求。
元素形态分析技术主要由样品采集、样品制备、分离/富集、定性/定量、分析报告等五部分组成。在整个形态分析过程中,样品制备过程是形态分析的关键环节,直接与分析结果的有效性息息相关。因此在样品制备过程中需要注意保持待测元素形态,同时避免污染,这使得样品制备过程较常规总量分析更加复杂和困难。因此,对操作人员提出了更高的要求,同时也延长了前处理时间。由于元素的某一形态,仅仅是元素总量的一部分,甚至是极少的一部分,因此对分析方法的灵敏度提出了更高的要求,只有高灵敏的检测技术才能满足元素形态分析的要求。此外,用于元素形态分析的标准物质和标准参考物还需要倚赖进口,在一定程度上影响了形态分析技术的推广。
因此,元素形态分析技术属于难度较高分析化学领域之一,对分析工作者以及相关分析仪器都要着较高的要求,目前仍部分处于实验室研究向例行常规分析转化的阶段,需要广大的分析工作者共同努力,推进这一技术的发展,满足国计民生各个领域的需求。
§张任-分析
第3楼2009/09/03
4\元素形态分析方法
1、差减法
早期的形态分析方法一般采用差减法进行测定,通过控制某些测量条件,实现总量和某些元素形态的测量,然后通过差减的方法得到其它元素形态的含量信息。例如通过测量总砷和三价无机砷,二者相减即可得到五价无机砷的浓度;如通过四价无机硒和总硒的测量,即可测得六价无机硒的含量。差减法相对比较简单,整个分析过程对实验条件的要求不高,但是该方法仅仅适用于元素形态较少的条件,操作较为繁琐,无法实现分析过程的自动化。
2、分别测定法
分别测定法是指首先利用物理或化学的分离方法对元素的各种形态进行有效分离后,再对其进行检测。例如在测定Cr(III)和Cr(VI)时,利用Cr(VI)在阴离子离子交换树脂吸附,而阳离子态的Cr(III)则不吸附的特性,将Cr(III)和Cr(VI)分离后,最后利用原子吸收法分别进行测定。该方法将元素形态的分离与测定分别进行,使得操作过程变得比较繁琐,同时在操作过程中可能会造成样品的损失以及元素形态的变化,对最终的测定结果产生比较大的影响。
3、联用技术
元素形态分析的通用原则是先对元素的各种形态进行有效分离,然后再逐一进行检测。近年来,人们在追求元素形态分析方法的高灵敏度、高选择性的同时,也一直在致力于提高分析过程的效率,缩短分析时间,力图实现整个分析过程的自动化。传统的元素形态分析方法将元素形态的分离与测定分别进行,操作过程繁琐,且会带来较多影响测定结果的不确定因素。
联用技术将高效的分离技术与高灵敏的检测技术有机结合,元素形态经过分离后,通过在线“接口”直接进入检测器进行检测,这样灵敏度、准确度和分析过程的效率都得到很大提高。联用技术是指将不同元素形态的分离与测定结合为一体,实现样品中元素不同形态的在线分离与测定。联用技术由于可以实现在线的有效分离与测定,因此已成为目前元素形态分析的主要手段。分离部分主要以高效的色谱和高效毛细管电泳分离手段为主,检测部分主要是高灵敏的光谱/质谱类检测器为主。此外联用技术还包括一些特殊的接口、特殊的样品引入技术、在线样品前处理技术以及能够控制联用技术多个功能单元的专用的工作站软件等。
§张任-分析
第4楼2009/09/03
5\联用技术的分离部分-01
联用技术的分离部分以色谱和高效毛细管电泳为主,此外还有利用衍生物的气化温度不同,通过逐步升温达到分离目的,如EPA1632方法用于测定水和组织中的砷形态,利用无机砷及甲基砷化合物与NaBH4反应后,衍生出沸点不同的AsH3(-56℃),CH3AsH2(-23℃),(CH3)2AsH(5℃),在液氮冷阱中被固化。然后开始由液氮(-195.8℃)冷却开始,逐步升温至室温,依据沸点的不同,沸点低的AsH3首先气化、逸出,在载气的携带下,分别在不同的时间到达检测器,最终达到分离检测的目的。
1.1毛细管电泳分离方法
电泳分离方法首先由瑞典化学家Arne Tiselius在20世纪30年代提出,用于血清中蛋白质的研究,并因此获得1948年的诺贝尔奖。电泳分离的原理在于带电粒子在直流电场的作用下,以不同速率运动,最终达到检测器的时间不同,而达到分离的目的。传统的电泳方法由于高电压引起的焦耳热,最终形成温度梯度、粘度梯度和速率梯度,造成区带展宽,最终影响到分离效率。在20世纪70-80年代,Virtanen、Jorgenson和Lukcas等人提出并发展了毛细管电泳(CE),由于截面积的减小而导致电泳介质的电阻值大大降低,有效抑制了焦耳热的生成,使得区带展宽效应得以有效控制,极大地提高了分离效率。毛细管电泳与传统的平板电泳法相比,具有分离效率高、分析速度快、样品(nL)及试剂用量少等优点。毛细管电泳根据分离机理的不同,可以分为毛细管区带电泳(CZE)、毛细管凝胶电泳(CGE)、毛细管等电聚焦(CIEF)、毛细管等速电泳(CITP)、毛细管胶束电动色谱(MECC)及毛细管电色谱(CEC)等六类。
毛细管电泳作为一种高效、快速分离技术,在元素形态分析领域具有很大的潜在应用优势。但是遗憾的是,其纳升级的进样量严重影响了分析的灵敏度,限制了在对分析灵敏度有着极高要求的形态分析领域中的应用;极低的流量(μL min-1)对检测器的接口提出了更高的要求,在雾化器、连接管路以及联用接口等方面均需要特殊设计,尤其在对因死体积造成的谱带展宽问题上的要求更为苛刻。目前,毛细管电泳作为联用技术的分离手段还仅仅停留在实验室研究阶段,真正作为成熟的分析手段用于日常的形态分析工作还为时尚早,但将来在与超高灵敏度的检测器联用时,将会具有极为广阔的应用前景。
1.2色谱分离方法
色谱法是现代分离分析科学中的一个重要分支,在近一个世纪的不断实践中,成为人们探索未知世界必不可少的工具,并逐渐发展形成一门独立的学科。色谱法的发展历史最早溯源于1901年,俄国植物学家M.S.Tswett首先提出利用层析法的原理分离植物色素,随后将这一方法命名为“色谱法”(Chromatography)。在这以后,色谱分离方法迅速被各国科学工作者应用和发展,并广泛应用于各种天然有机化合物的分离与分析。20世纪30-40年代,发展了柱分配色谱、纸色谱;50年代发展了气相色谱法和薄层色谱法;60年代发展了凝胶色谱法和高效液相色谱法;70年代发展了高效毛细管气相色谱法,之后又发展了高效毛细管电色谱;90年代又出现了光色谱。
色谱法利用物质在两相(气/固,液/固等)中吸附或分配系数的差异达到分离的目的。当两相做相对移动时,物质在两相间进行多次反复的分配,使得原来微小的分配差异得到放大,最终能够将复杂的混合物组分逐一分离,分别检测。色谱分离的必要条件是多种待分离组分在性质上存在微小差别,这也是色谱分离的根本;待分离组分在两相间进行了上千次甚至是上百万次的交换,则是色谱分离的充分条件。色谱分离的机理如图1-1所示,含有ABC三种组分的混合物在流动相的携带下,经过与固定相的不断作用,最终以ACB的先后顺序分离开。根据保留时间的不同,即可实现ABC三个组分的定性分析;对多个组分分别进行峰高或峰面积的定量校正,即可实现多个组分的定量计算。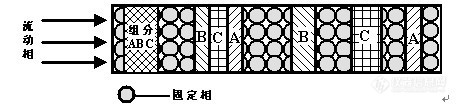
§张任-分析
第5楼2009/09/03
5\联用技术的分离部分-02
色谱法经过近百年的发展,形成多个分支,在联用技术中应用较为广泛的主要有高效液相色谱法(HPLC)和气相色谱法(GC)等。
1.2.1高效液相色谱法(HPLC)
高效液相色谱法以液体为流动相,利用各组分在流动相和固定相(色谱柱填料)之间的分配能力的差异而实现分离分析的柱色谱技术。高效液相色谱法具有分离效率高、选择性强以及分析速度快等优点,适用于80%以上物质的分离分析,是目前在色谱法中发展最快、应用最广泛的分离技术,也是在形态分析领域应用最为广泛的分离手段。高效液相色谱法适用于高沸点、不易挥发、热不稳定以及分子量较大的物质,在形态分析中主要用于小分子的有机金属化合物和大分子的金属缔合物如金属硫蛋白(MT)等物质的分离。
用于形态分析的HPLC法主要有以下几种类型:
(1)反相色谱法(RP-HPLC)
反相色谱法是以非极性表面的载体作为固定相,以比固定相极性强的溶剂作为流动相的一种液相色谱分离模式。反相色谱法的固定相一般为表面键合疏水基团(C4、C8、C18或苯基等)的高纯硅胶微球或者聚合物材料,流动相一般为甲醇或乙腈与水的混合物,此外还有缓冲盐和离子对试剂等。反相色谱法是目前液相色谱中应用最为广泛的分离模式,在流动相组成改变时,流动相中的强有机溶剂能够迅速在固定相表面达到平衡,因此特别适合于梯度洗脱;此外,在反向色谱中,溶质在固定相上的保留是基于分子间的非特异性疏水作用,由于大部分待测物都含有疏水基团,能够与固定相产生相互作用,因此反向色谱法成为理想的、普遍适用的方法。在反向色谱法中,含水的流动相更适于一些不溶于有机溶剂,同时又会与极性固定相发生强烈吸附作用的极性化合物。在生物大分子、蛋白质以及金属结合的生物大分子的分离分析方面,已经受到越来越多的关注。反相色谱法可用于从离子型到非离子型化合物的分离;固定相化学性质稳定,分离的重复性较好;分析速度快,且平衡时间短。对于应用最为广泛的硅胶基质的键合固定相,一般适用的pH值限于2~7.5之间,且表面的硅羟基会严重影响色谱分离效果。
对元素形态分析而言,一些中性有机金属化合物较为适合使用反向色谱法直接分离,如有机锡化合物、有机硒化合物和有机锰化合物等。如无机锰和有机锰的分离可以在流动相组成为甲醇/水-醋酸缓冲液体系中,在C18柱上实现分离;有机硒的分离可以在流动相组成为甲醇/水-甲酸铵缓冲液体系中,在C18柱上实现分离;有机锡的分离可以在流动相组成为水/三氟乙酰酸-甲酸缓冲液体系中,在C18柱上实现分离。
(2)反相离子对色谱法(RPIP-HPLC)
反相离子对色谱法是将与待分离离子带相反电荷的离子(对离子或反离子)加入到流动相中,与待分离离子形成弱极性离子对,该离子对在流动相中不易离解而迅速与键合相作用,进而在流动相和固定相之间进行多次分配,最终实现分离的一种色谱模式。离子对色谱法发展于20世纪70年代初,分为正相和反相离子对色谱法,目前主要使用的是反相离子对色谱法。反相离子对色谱法与反相色谱法不同的是流动相中加入了一种与待分离物质电荷相反的物质(离子对试剂),所使用的固定相则与反相色谱法类似,多为长碳链烷基(C8或C18)。离子对试剂的种类和浓度对分离效果都有较大的影响,选择离子对试剂一般基于以下原则:如果被分离物质是强酸或强碱,离子对试剂可以是强碱、弱碱或强酸、弱酸;如果被分离的物质是弱酸或弱碱,则选用的离子对试剂必须是强碱或强酸。常用的离子对试剂为长链的烷基磺酸盐或烷基铵盐。一般阴离子型的离子对试剂,只会使样品中的阳离子保留在色谱柱上,阴离子在死体积流出;而采用阳离子型的离子对试剂则会得到相反的结果。疏水性较强的离子对试剂,浓度在4~10mmol L-1较为适宜;疏水性较弱的离子对试剂,浓度在10~40mmol L-1较为适宜。pH值是影响保留值和选择性的最重要的因素之一,对于酸性物质,当pH>pKa时,将以离子状态存在,此时离子对试剂对保留值发挥了极其重要的作用;而当pH≤pKa时,将以中性状态存在,离子对试剂对保留值几乎没有发挥作用。pH值对碱性物质的影响则恰好相反。因此在RPIP-HPLC中,选择合适的缓冲液,使待分离物质以所需要的电荷状态存在是实现色谱分离的先决条件。值得注意的是,在特定的pH值下,某些组分没有完全解离,酸碱平衡的慢动力学过程可能会引起色谱峰的分裂和展宽。
反相离子对色谱法由于适用的范围广,在元素形态分析领域也成为主要的分离手段之一。在分离阴离子型的砷形态时,普遍采用季铵盐型的离子对试剂;而在分离阳离子型的砷形态时,普遍采用阴离子型的离子对试剂,如烷基磺酸盐等。在分离阳离子态的无机汞和有机汞时,可以采用四丁基溴化铵、溴化双十二烷基二甲铵等离子对试剂。
反向离子对色谱法兼有反向色谱法和离子交换色谱法的特点,保持了反向色谱法操作简便、分离柱效高等优点,同时能够分离离子型和中性样品,可以认为是离子交换色谱和反向色谱的拓展和延伸。
(3)离子交换色谱法(IE-HPLC)
离子交换色谱法是元素形态分析领域应用最为广泛的色谱分离手段。样品离子和键合在固定相(树脂或硅胶)上的带电荷的官能团之间发生离子交换,由于不同的离子与固定相之间的作用力不同,作用力弱的离子不易保留,首先被洗脱,而作用力强的离子则最后被洗脱,最终达到分离的目的。离子交换色谱的固定相上的交换基团可以带正电荷也可以带负电荷,因此可以分为阴离子交换色谱和阳离子交换色谱两种。
阴离子和中性的砷形态可以通过阴离子交换色谱法进行分离,阳离子砷形态和汞形态则可以通过阳离子交换色谱法分离;无机硒和硒的氨基酸配合物也可以利用阴离子交换色谱进行分离。
1.2.2气相色谱法(GC)
气相色谱法以气体作为流动相,利用各组分与固定相之间相互作用力的差异,进行分离分析的色谱方法。与液相色谱法不同的是气体作为流动相一般仅起到携带作用,并不与待分离组分发生作用。根据固定相的类型,气相色谱法又分为气/固和气/液两类,一般指应用最广泛的气/液色谱法。用于气相色谱法的色谱柱分为填充柱和毛细管柱两种,前者装置比较简单,分析成本较低,但分辨率不高,针对较为复杂的组分难以达到令人满意的分离结果;毛细管柱能够提供理想的分辨率和分离度,分析速度快且峰形更为尖锐,可以获得更高的灵敏度,已经成为气相色谱法的首选色谱柱。毛细管柱的柱容量有限,仅有几微升,影响到分析的灵敏度,最终限制了其在一些超痕量分析中的广泛应用。
气相色谱法用于元素形态分析的主要目标分析物为挥发性或热稳定性的物质或可以通过衍生反应转变为挥发性或热稳定的物质,在形态分析中的研究重点主要是有机汞、有机铅和有机锡化合物等沸点较低且热稳定性较高的元素形态.
(未完待续)

































